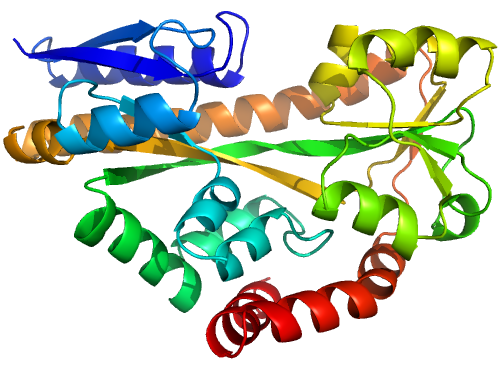
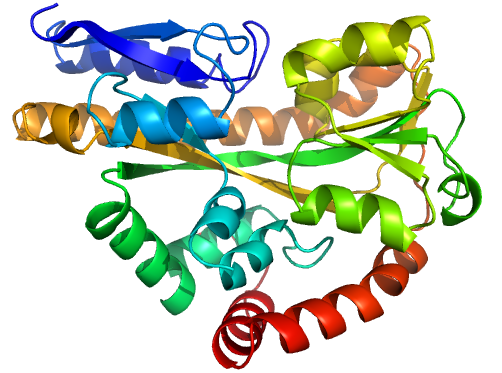
Figure 1 (stereo). Illustrations of sequence-identical chains, specifically the open (2cex_A, left) and closed (3b50_A, right) forms of the SiaP TRAP SBP (Fischer et al., 2010), rainbow-coloured along the chain.
, Marcus Fischer and Garib N. Murshudov
MRC Laboratory of Molecular Biology, Hills Road, Cambridge CB2 0QH
Email: nicholls@mrc-lmb.cam.ac.uk
"Procrustes owned two beds, one small, one large; he made short victims lie in the large bed, and the tall victims in the short one…" (Taleb, 2010)
Procrustes was a mythological Greek villain whose victims were stretched and cut in order to fit the shape of his bed. The "Pro" in ProSMART is due to its use of Procrustes analysis (Gower, 2010; Gower and Dijksterhuis, 2004; Catell and Hurley, 1962) for comparing local regions of structure between two protein chains. The name is fitting in this context due to manipulating the coordinates from one structure in order to optimally fit those in another, efficiently achieving a measure of local main-chain r.m.s.d. at the chosen level of structural resolution. By performing an exhaustive structural comparison of all local regions between two input structures, ProSMART is able to produce alignments by optimising the net agreement of local structures along the chain. The resulting alignment is thus independent of the global conformation of the compared chains - this is subsequently exploited for various purposes…ProSMART (Procrustes Structural Matching Alignment and Restraints Tool) has two main purposes: conformation-independent comparison of protein structures, and the generation of interatomic distance restraints for subsequent use in macromolecular crystallographic refinement by REFMAC5 (Murshudov et al., 2011, 1997; Nicholls et al., 2012). Therefore, the tool comprises two major components:
ProSMART is written in C++, takes one or many PDB files as input, and can be run from the command line or using a CCP4i (Potterton et al., 2003) interface (see below). Mac/Linux and Windows versions are available.
This article gives a brief overview of some of the features available in ProSMART, a discussion regarding the generation of external structural restraints, an overview of the ways to run ProSMART, and the nature of the output.
Several features are available for performing different types of comparative structural analyses, depending on the level of structural similarity of the compared protein chains. ProSMART is particularly well suited to the comparative analysis of homologous chains in different global conformations, e.g. apo versus holo. Further to being able to achieve an alignment between similar structures, you could in principle use ProSMART to create an optimal alignment between completely dissimilar structures. The alignment achieved by ProSMART is effectively the optimal conformation-independent net agreement of local structures along the chain; alignment filtering according to local structural dissimilarity scores may be subsequently performed, if desired.
Further to achieving a conformation-independent alignment, ProSMART automatically performs identification and superposition of rigid substructures that are conserved between the compared chains.
ProSMART uses various residue-based scores for describing the dissimilarity of aligned residues' local structural environments. These scores include measures that are robust, allowing the identification of similarity in the presence of conformational change. Other scores are very sensitive, being able to detect subtle changes in the local structural environments that would otherwise be extremely hard to detect. These scores complement each other, maximising the amount of information achieved when performing such structural analyses (see Figure 2).
Another major feature of ProSMART is that the default generated output allows publication-quality illustrations to be quickly and easily achieved using the molecular graphics software CCP4mg (McNicholas et al., 2011) or PyMOL (Schrödinger; DeLano, 2002), for various types of comparative structural analyses. For example, Figures 2-5 display default results from ProSMART in PyMOL, without any subsequent manipulation of superpositions or colouring (the illustrations were not tediously prepared by hand!).
The structural comparison methods used in ProSMART will be described in a future article (in the meantime, details of the methods are described by Nicholls, 2011). For the purposes of this article, we shall provide a few figures to briefly illustrate some of the functionalities that may be applied to highly homologous chain-pairs (thus would be of most relevance to cases involving external restraint generation).
Figure 1 (stereo). Illustrations of sequence-identical chains, specifically the open (2cex_A, left) and closed (3b50_A, right) forms of the SiaP TRAP SBP (Fischer et al., 2010), rainbow-coloured along the chain.
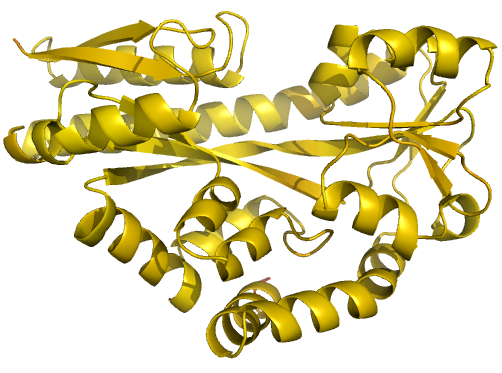
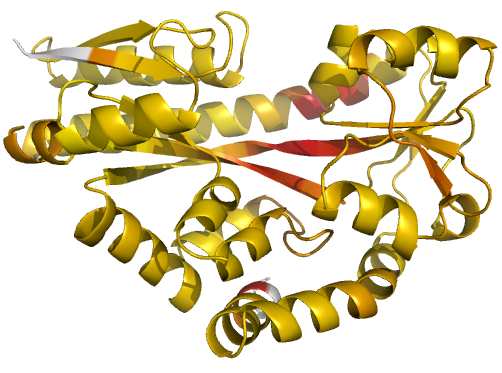
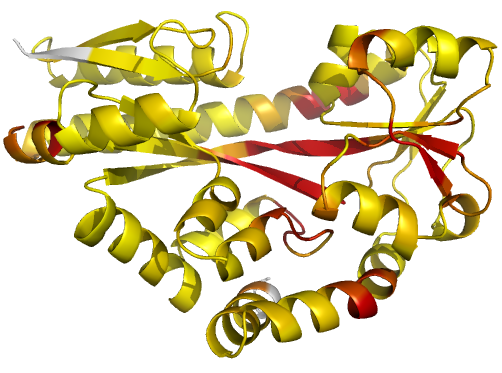
Figure 2 (stereo). Illustrations of simple results from the default ProSMART comparison of 2cex_A and 3b50_A, coloured using a gradual colour gradient according to main-chain dissimilarity scores (yellow implies similarity, red relative dissimilarity). For clarity, only the chain 2cex_A is shown (if shown, residues in 3b50_A would have been coloured the same as the corresponding residues in 2cex_A). These depictions allow quick visual identification of exactly which regions are structurally very similar, and which exhibit differences. The "minimum score" (left) is highly insensitive to global conformation - note that all residues are aligned and identified as very similar despite the global conformational change. The "central score" (middle) is more sensitive to differences in local structural environment - note that locally distorted regions such as the hinge are easily identified. The "intrafragment rotational dissimilarity score" (right) is sensitive to curvature and torsion of the local backbone - this score is useful for identifying regions that exhibit subtle backbone deformations that can be very hard to otherwise identify or quantify.
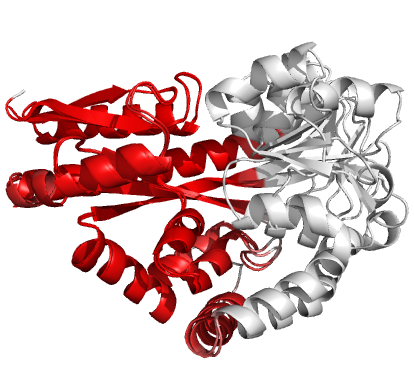

Figure 3 (stereo). Superpositions arising from the rigid substructure identification results from default ProSMART comparison of 2cex_A and 3b50_A, coloured according to cluster scores. Two rigid substructures identified, coloured red (left) and green (right), corresponding to the two domains.
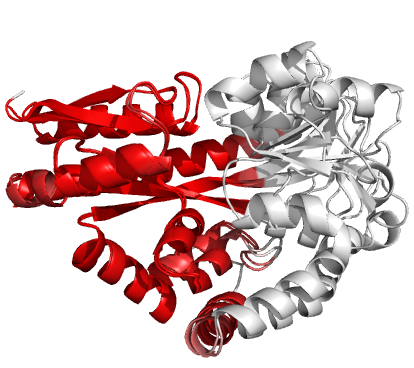
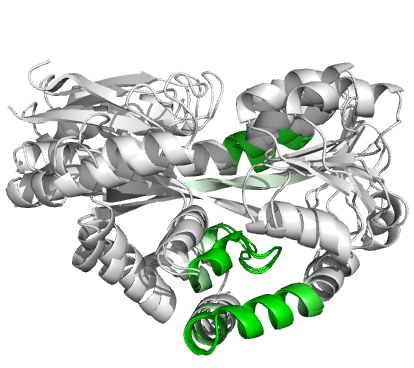
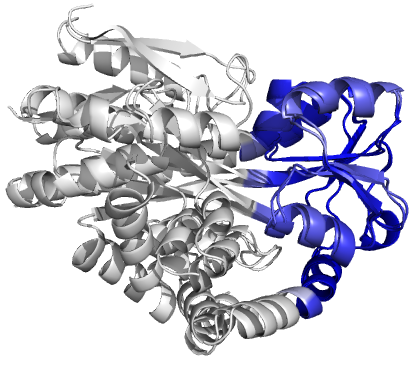
Figure 4 (stereo). Superpositions arising from the rigid substructure identification results from the ProSMART comparison of 2cex_A and 3b50_A using a fragment length of 7 residues (keyword: -len), coloured according to cluster scores. In contrast with Figure 3, which used the default fragment length of 9 residues, three rigid substructures are identified, coloured red (left), green (middle) and blue (right). These substructures correspond to two domains and the hinge region. This helps to illustrate the utility of performing comparative analyses at multiple levels of structural resolution.
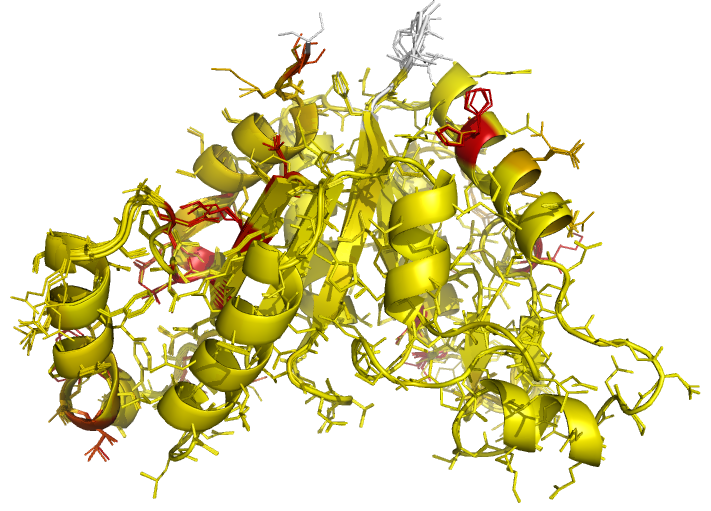
Figure 5 (stereo). Illustrations of superposed NCS-related chains in the structure of BioD with PDB code 3MLE (Porebski et al., 2012). Residues are coloured according to side-chain RMSD relative to the local coordinate frame, allowing easy visual identification of residues with side-chains in different conformations. This can be particularly useful for cross-validation during various stages of the refinement process (e.g. by identifying changes in side-chain conformation before/after refinement, and identifying which side-chains are/aren't pulled towards reference structures after application of external side-chain restraints).
Information from well-refined higher-resolution structures may be used to improve reliability of low-resolution structures during refinement, provided that the reference structure is sufficiently similar to the target. When using such external information, there are various questions one should ask that might affect restraint generation, for example:
Here, we discuss how ProSMART can be used to generate interatomic distance restraints for subsequent use in refinement by REFMAC5. Details of the methods used, along with examples of application, are described in an upcoming article (Nicholls et al., 2012).
External reference structures would usually be identical or close homologues, although in theory any structures could be used. There are no hard-coded limits on sequence identity, although intuitively only sufficiently similar structures should be used as external information. However, ProSMART is general and flexible, and provides the ability to allow user-input beyond the realms of common sense!
It is possible to use ProSMART to generate restraints using multiple reference structures. If multiple homologues are provided then REFMAC automatically selects the regions that are most consistent with the existing structure, only using the restraints that are closest to the current interatomic distances. This is done separately for each interatomic restraint. This means that if many structures are used as references, only the structure(s) most similar to the target should actually affect refinement, in theory. Nevertheless, we do not suggest that blindly using all homologues is a good strategy - manual consideration and common sense should always prevail! Carefully selecting one or a selection of homologous structures that are highly conserved in local structure would generally be a better approach, at present.
Note that the target and reference structures should be conserved in local structure - conservation of overall fold at the global level is neither necessary nor sufficient. To clarify, the conformation-independent approach of ProSMART means that sensible external restraints may be generated even if the target and reference structures adopt different global conformations (e.g. apo and holo forms). Indeed, the external restraints generated by ProSMART always operate locally, and thus they should not enforce global rigidity.
If a reference structure contains multiple (e.g. NCS-related / multimeric) chains in the PDB file then ProSMART will generate restraints for all target chains using information from all reference chains, by default. This may be desirable in some cases, for reasons outlined above. However, it is also possible to avoid this, and instead generate restraints only for the chain considered to be the best match to the target chain, in terms of net local structural similarity (keyword: -restrain_best).
It is important not to forget that reference structures may contain errors - the naïve application of restraints from such structures may cause errors to propagate into the target structure during refinement. Consequently, it is recommended to inspect reference structures, and possibly also consider their re-refinement before use, e.g. using PDB_REDO (Joosten et al., 2009).
It is recommended to perform a comparative structural analysis between target and reference structures prior to refinement (i.e. viewing the results from ProSMART ALIGN). This allows the user to visualise and quantify local (dis)similarities between the structures and thus make better-informed decisions regarding the local structural similarity of their presumed homologue to the target, prior to the application of the external restraints. It is also recommended for such comparative analyses to be performed following refinement, allowing the user to visualise and quantify the effect of the external restraints on local structure. In tandem with inspection of the electron density, this would help in deciding whether the external restraints were constructive (thus should be kept) or destructive (thus should be removed or replaced) in each local region, and also help in deciding the values of REFMAC5 external restraint weighting parameters (see below).
Ideally, the target and reference structures should be manually inspected, and the decision should be made as to whether restraints should or should not be generated for all regions. Challenging structures may require special attention, however, this should be deemed worth the effort when the alternative is to produce a better diffracting crystal. If there are some regions that are actually different between the two structures, and it is decided that external restraints should not be generated for these regions, then the restraints corresponding to these residues/regions can be removed (keywords: -restrain and -restrain_rm).
Additionally, it is possible to specify that restraints should only be generated for regions that are sufficiently conserved, in terms of local main-chain (keyword: -cutoff) and/or side-chain (keyword: -side_cutoff) similarity.
By default, restraints are generated for all aligned portions of structure, regardless of local structural conservation - the user must decide whether dissimilarity thresholds are appropriate in the particular case.
Alternatively, PDB files may be separately filtered in a way deemed appropriate for subsequent restraint generation, either manually or using a tool such as Tim Grüne's mrprep (Grüne, 2012).
Note that restraints can be generated only for main-chain chain atoms (keyword: -main), or for both main-chain and side-chain atoms (keyword: -side).
At present, caution is due when optimising certain parameters in REFMAC5, most notably the external restraints weight (keyword: EXTERNAL WEIGHT SCALE) and Geman-McClure parameter (which down-weights outliers, keyword: EXTERNAL WEIGHT GMWT), in order to successfully apply external restraints during refinement. For more information, see Nicholls et al. (2012). Information on how to provide REFMAC5 with such keywords can be found here and here; they can also be specified using the CCP4i REFMAC5 GUI (see below).
Appropriate values for these parameters will vary for each case, and will depend on many factors. Such factors may include properties of the target structure's refinement in the absence of external restraints (e.g. X-ray resolution, quality of electron density, geometry weight, number of NCS-restrained chains) and also properties of the external structural information, such as the quality of the external information (i.e. reference structures), and the type of external restraints (e.g. main-chain, side-chain, generic SS H-bond or fragment-based restraints), etc.
As an aside, it is important to make sure that sufficient cycles of REFMAC5 are executed when using external restraints - the external restraints will seem to have little effect if running only 5 refinement cycles. Something like 20-30 cycles may be required, and even more if also using jelly-body restraints (Murshudov et al., 2011).
Further to generating restraints using homologous protein structures, ProSMART allows the generation of restraints to specific n-residue structural fragments (keyword: -lib to use all fragments present in the local library). Such generic fragment-based restraints can be used to help the structure better-adopt a desired conformation, for example, using an ideal helical fragment to help stabilise the formation of a helix. The implemented approach is generalised, allowing any structures to be used as reference fragments in principle (e.g. the user may provide their own fragment coordinate files). At present, ProSMART comes with fragments representing an ideal alpha-helix (keyword: -helix) and a representative beta-strand (keyword: -strand), which can be used to generate in-sequence quasi-secondary-structure restraints. Note that these restraints are not considered true secondary-structure restraints, since they restrain all atom-pairs in the local structural environment, rather than just those considered to be hydrogen-bonded (also, note that the residue alignment is not assigned using a traditional method such as DSSP). This method is considered powerful, since it does not require the low-resolution structure to be sufficiently well modelled to assign secondary-structure using hydrogen-bonding patterns. For example, you may want to use helix restraints from ProSMART to force a particular troublesome helix to maintain a reasonable helical conformation during early/intermediate stages of model building/refinement.
Further to existing functionalities, generic restraints representing specific atomic interactions, notably hydrogen bonds (e.g. in alpha-helices, across beta-sheets), and also external restraints for DNA/RNA from homologous structures, will be available imminently in an upcoming version of ProSMART (which should be available by the time you read this!).
Please contact the authors for any assistance or information (see here).
Basic instructions on how to run ProSMART using a command line interface can be found here.
Typing prosmart will give a list of all keywords in that version. A list of keywords with more detailed descriptions can be found here. Note that keywords can be specified in an external text file (keyword: -f), which can be useful if the arguments list gets too long.
Instructions on how to use a command line interface to run REFMAC5 with external restraints generated by ProSMART can be found here.
An updated version of the REFMAC5 CCP4i GUI, which includes the ability to automatically run ProSMART for external restraint generation, has been developed by Martyn Winn at STFC Daresbury. The updated interface is currently available, and will be distributed in the next release of the CCP4 suite (version 6.3). The REFMAC5 GUI allows you to specify for ProSMART to be automatically run (using mainly default settings) prior to running REFMAC5 with the generated external restraints. Note that this new feature will be hidden if ProSMART is not installed.
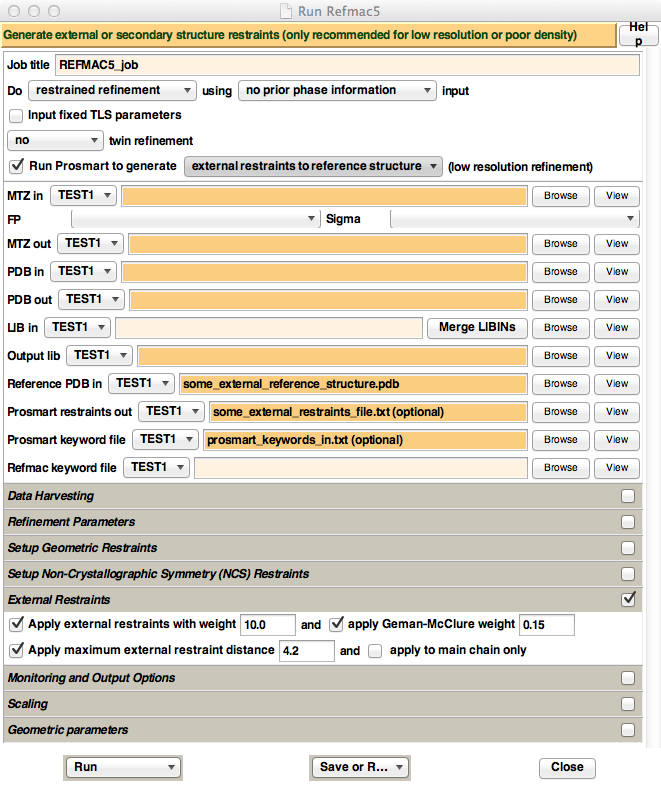
Figure 6. Appearance of the new REFMAC5 CCP4i GUI, when ProSMART is installed. For illustrative purposes only, text has been inputted into the "Reference PDB in", "Prosmart restraints out", and "Prosmart keyword file" fields - these are the three fields that appear when enabling the "Run Prosmart to generate" option. Also, the "External Restraints" tab has been opened, which contains options that control the way REFMAC5 uses the external restraints generated by ProSMART.
To automatically generate and use ProSMART external restraints, make sure that the "Run Prosmart to generate" button is ticked. This will cause three input file options to appear: "Reference PDB in" (analogous to the -p2 keyword), "Prosmart restraints out", and "Prosmart keyword file" (analogous to the -f keyword). For simple execution, only the "Reference PDB in" has to be specified. For more advanced functionality, create a simple text file containing the desired ProSMART keywords, and pass this file to the GUI in the "Prosmart keyword file" field.
The REFMAC5 GUI has four options controlling behaviour of the external restraints during refinement, namely the external restraints weight, Geman-McClure parameter, maximum external restraints distance, and whether or not side-chain atoms are to be restrained in addition to main-chain atoms. These options can be found in the "External Restraints" tab in the GUI. Unless there is a very good reason for doing otherwise (or just want to experiment!), it is highly recommended to enable "Apply maximum external restraint distance", and set it to 4.2 (which is a magic number that tends to always be approximately optimal). Other parameters should be experimented with; see Nicholls et al. (2012) for more information.
As an aside, it may be advisable to select "Run&View Com File" instead of "Run" from the bottom-left drop-down box. This will display the command (with keywords) used to run REFMAC5 before actual execution - this will confirm whether REFMAC5 is being run as intended!
To ensure that CCP4i is running ProSMART and REFMAC5 as intended, it is recommended to inspect the output log file to see exactly what command was used to run ProSMART, and confirm that the ProSMART job completed successfully. The log file can be accessed from the main CCP4i GUI by double-clicking the appropriate job (or alternatively selecting "View Files from Job" then "View Log File"). For example, at the top of this log file you should see something like this:
***************************************************************************
* Information from CCP4Interface script
***************************************************************************
*** Starting Prosmart to determine restraints to external structure ***
Using command: prosmart -p1 "some_pdb.pdb" -o "some_location" -side -p2 "another_pdb.pdb"
Writing results to directory "some_directory"
***************************************************************************
This is then followed by the main ProSMART log. Upon successful completion, the following lines (or similar) will be displayed after the ProSMART log:
***************************************************************************
* Information from CCP4Interface script
***************************************************************************
*** Prosmart finished ***
Copying file of restraints some_file.txt to another_file.txt
***************************************************************************
---------------------------------------
Standard External All
Bonds: 7711 22212 29923
Angles: 13788 0 13788
Chirals: 706 0 706
Planes: 1372 0 1372
Torsions: 3296 0 3296
---------------------------------------
Usage of the external restraints is confirmed by the fact that the number of external bonds is non-zero (and generally quite large - in this case, 22212).
As of REFMAC5 version 5.7.0022, more output regarding external restraints will be available in the log file, providing information about the input parameters and options used. This information will look something like this (located above the standard restraints table):
--------------------------------------------------------------------------------
External restraints group : 1
External restraints file :input_keywords
Fail if one of the atoms involved in the restraints is missing in the pdb file
Use restraints for all defined atoms
Ignore restraints if abs(dmod-drest) > 50.000000 *sigma
Ignore retraints if input dist > 1.00000003E+32
Weight scale sigmas : 1.0000000
Weight min sigma : 0.0000000
Weight max sigma : 100.00000
GM parameter : 0.10000000
Number of distances : 10809
Number of angles : 0
Number of torsions : 0
Number of planes : 0
Number of chirals : 0
Number of intervals : 0
--------------------------------------------------------------------------------
It is often desirable to run ProSMART separately (e.g. from the command line, or the ProSMART GUI) instead of running ProSMART automatically using the REFMAC5 GUI. In this case, it is necessary to provide the REFMAC5 GUI with the external restraints file generated by ProSMART. This restraints file should be provided to the GUI in the "Refmac keyword file" field. Note that, in this case, the "Run Prosmart to generate" button should not be ticked.
For more control over how REFMAC5 deals with the external restraints, it is advised to create a REFMAC5 external keywords file that specifies the location of the ProSMART restraints file, and then pass this keywords file to the GUI using the "Refmac keyword file" field (latest versions of REFMAC5 only). To do this, create a simple .txt file (note: must be plain text, not rich text) that contains the commands to tell REFMAC5 to use the ProSMART restraints file. For example, this file may simply contain the line:
@my_prosmart_restraints_file.txt
where my_prosmart_restraints_file.txt is the name/location of the restraints file generated by ProSMART. The @ symbol specifies for REFMAC5 to parse the my_prosmart_restraints_file.txt file and use any external restraints found.
In practical application, it is necessary to tell REFMAC5 how to deal with these restraints, e.g. what weighting parameters to use. This can be achieved by specifying EXTERNAL keywords. For example, the options enabled in the "External Restraints" tab in the GUI shown in Figure 6 would be specified:
EXTERNAL DMAX 4.2
EXTERNAL WEIGHT SCALE 10
EXTERNAL WEIGHT GMWT 0.15
@my_prosmart_restraints_file.txt
Note that any EXTERNAL keywords must be specified above the @my_prosmart_restraints_file.txt line.
In addition, the EXTERNAL USE MAIN keyword can be used to discard any side-chain restraints that may be present in the external restraints file (this equivalent to the "apply to main chain only" option in the REFMAC5 GUI). Of course, regardless of whether this keyword is specified, side-chain atoms will only be restrained if you tell ProSMART to generate side-chain restraints (keyword: -side). The EXTERNAL USE ALL keyword may be specified to ensure that all external restraints present in the ProSMART restraints file are used.
As of REFMAC5 version 5.7.0022, an additional keyword EXTERNAL USE HBOND may be specified, which only uses external restraints that may form hydrogen bonds (i.e. restrained atom-pair are donor and acceptor); all other restraints present in the input file will be ignored. Note that only one of EXTERNAL USE MAIN, EXTERNAL USE ALL, and EXTERNAL USE HBOND may be specified (per external restraints file).
For clarification of the format of REFMAC5 keywords files, here is an example keyword file.
A ProSMART GUI for CCP4i is available from the Murshudov Group website (under Software→ProSMART→Download ProSMART). Installation instructions can be found here. This GUI provides a user-friendly alternative to running ProSMART from the command line, providing near-comprehensive functionality for both ProSMART ALIGN (structural comparison) and ProSMART RESTRAIN (external restraint generation) features. Note that restraints generated using this GUI can be passed to the REFMAC5 GUI via an external keywords file, as described above.
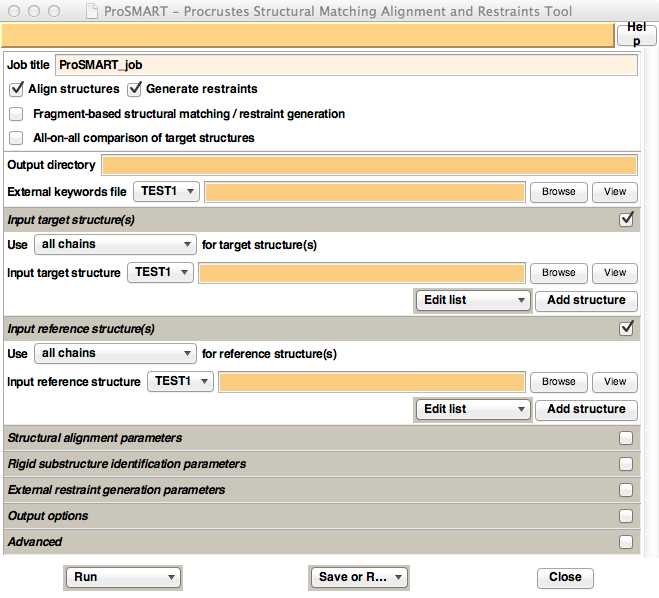
Figure 7. Appearance of the ProSMART CCP4i GUI.
After running ProSMART, there are two major sources of general results information: the main ProSMART log file, and the ProSMART HTML output page.
The main ProSMART log file indicates what options were used (e.g. whether or not side chain restraints were generated), which chain-pairs were considered, and whether the job finished successfully. In the CCP4i GUIs, this log file can be accessed by double-clicking the appropriate job (or alternatively selecting "View Files from Job" then "View Log File"). If using a command line interface, this information is printed to screen.
The ProSMART HTML output page allows navigation of all major results files, provides an easy way of viewing the log files from all pairwise executions of ProSMART ALIGN and ProSMART RESTRAIN, and provides a list of the program options used (which can be useful for future reference). The HTML results page may be accessed from the ProSMART CCP4i GUI under "View Files from Job". Otherwise, this results file may be found in the ProSMART output directory (which by default would be here: ProSMART_Output/ProSMART_Results.html relative to the current working directory).
The major output files generated by ProSMART that can currently be viewed from the HTML output page include:
The output superposed PDB files may be viewed using PyMOL (Schrödinger; DeLano, 2002). The output PyMOL colour scripts may be used to colour residues according to their various dissimilarity scores (including those illustrated in Figures 2-5).
The latest release of CCP4mg (McNicholas et al., 2011) includes an experimental ProSMART analysis feature that allows results from a ProSMART execution to be loaded for a powerful interactive illustration of the comparative analyses. Unlike with PyMOL, CCP4mg does not use/require ProSMART to provide superposed PDB files and colour scripts in order to achieve the desired effect. The ProSMART transformation files are used to superpose both global alignments and any identified rigid substructures; these structures may be coloured according to their residue-based dissimilarity scores, with colour gradients altered in real-time using a slider.
ProSMART will be distributed as part of the CCP4 suite (Winn et al., 2011) in the upcoming release (version 6.3). Latest versions, along with simple installation instructions and documentation, are always available from the Murshudov Group website: http://www2.mrc-lmb.cam.ac.uk/groups/murshudov/ (under Software→ProSMART).
For more information, please contact: nicholls@mrc-lmb.cam.ac.uk. Any comments or questions are always welcome!
This article may be freely cited and referenced, although it is preferred that references to restraint generation using ProSMART be made to Nicholls et al., 2012.
We would like to thank Martyn Winn for developing the REFMAC5 GUI, Stuart McNicholas for working on the ProSMART analysis features in CCP4mg, Marcin Wojdyr and Charles Ballard for providing advice on software and installation issues and working on the integration of ProSMART into CCP4, and CCP4 for support and distribution. We would also like to thank our colleagues for interesting discussions, and the many users who have provided useful feedback, resulting in greatly improved functionality.
This work was supported by the Medical Research Council (grant number: MC US A025 0104). Part of this work was carried out whilst the authors were at the Structural Biology Laboratory, Department of Chemistry, University of York, during which time RAN was funded by a BBSRC Ph.D. Studentship, MF was funded by a Wild Fund Scholarship and a BBSRC Ph.D. Studentship, and GNM was funded by the Wellcome Trust.
Catell, R.B. and Hurley, J.R. (1962) The Procrustes program in producing direct rotation to test a hypothesized factor structure. Behavioural Science 7, 258-262.
DeLano, W.L. (2002) The PyMOL Molecular Graphics System.
Fischer M., Zhang Q.Y., Hubbard, R.E. and Thomas G.H. (2010) Caught in a TRAP: substrate-binding proteins in secondary transport. Trends in Microbiology 18(10), 471-478.
Gower, J.C. (2010) Procrustes methods. Wiley Interdisciplinary Reviews: Computational Statistics 2(4), 503-508.
Gower, J.C. and Dijksterhuis, G.B. (2004) Procrustes problems. Oxford University Press, USA.
Grüne, T. (2012) mrprep - PDB preparation tool for use with ProSmart or for Molecular Replacement. http://shelx.uni-ac.gwdg.de/~tg/research/programs/mrprep/
Joosten, R.P., Salzemann, J., Bloch, V., Stockinger, H., Berglund, A.C., Blanchet, C., Bongcam-Rudloff, E., Combet, C., Da Costa, A.L., Deleage, G., Diarena, M., Fabbretti, R., Fettahi, G., Flegel, V., Gisel, A., Kasam, V., Kervinen, T., Korpelainen, E., Mattila, K., Pagni, M., Reichstadt, M., Breton, V., Tickle, I.J., Vriend, G. (2009) PDB_REDO: automated re-refinement of X-ray structure models in the PDB. Journal of Applied Crystallography 42(3), 376-384.
McNicholas, S., Potterton, E., Wilson, K. S. and Noble, M. E. M. (2011) Presenting your structures: the CCP4mg molecular-graphics software. Acta Crystallographica D67, 386--394.
Murshudov G.N., Skubak P., Lebedev A.A., Pannu N.S., Steiner R.A., Nicholls R.A., Winn M.D., Long F. and Vagin A.A. (2011) REFMAC5 for the Refinement of Macromolecular Crystal Structures. Acta Crystallographica D67, 355-367.
Murshudov G.N., Vagin A.A. and Dodson E.J. (1997) Refinement of Macromolecular Structures by the Maximum-Likelihood Method. Acta Crystallographica D53, 240-255.
Nicholls R.A. (2011) Conformation-Independent Comparison of Protein Structures. University of York (thesis). http://etheses.whiterose.ac.uk/2120/.
Nicholls R.A., Long F. and Murshudov G.N. (2012) Low resolution refinement tools in REFMAC5. Acta Crystallographica D68, 404-417.
Porebski P.J., Klimecka M., Chruszcz M., Nicholls R.A., Murzyn K., Cuff M.E., Xu X., Cymborowski M., Murshudov G.N., Savchenko A., Edwards A. and Minor W. (2012) Structural characterization of Helicobacter pylori dethiobiotin synthetase reveals differences between family members. FEBS Journal.
Potterton E., Briggs P., Turkenburg M. and Dodson E. (2003) A graphical user interface to the CCP4 program suite. Acta Crystallographica D59, 1131-1137.
Schrödinger, LLC. The PyMOL Molecular Graphics System, Version 1.5.0.1.
Taleb, N.N. (2010) The bed of Procrustes: philosophical and practical aphorisms. Random House.
Winn, M.D., Ballard, C.C., Cowtan, K.D., Dodson, E.J., Emsley, P., Evans, P.R., Keegan, R.M., Krissinel, E.B., Leslie, A.G.W., McCoy, A., McNicholas, S.J., Murshudov, G.N., Pannu, N.S., Potterton, E.A., Powell, H.R., Read, R.J., Vagin, A. and Wilson, K.S. (2011) Overview of the CCP4 suite and current developments. Acta Crystallographica D67, 235-242.