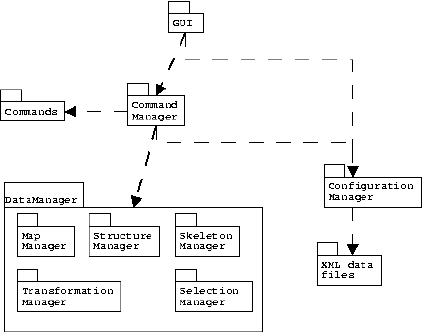
| Figure 4: UML (Universal Modeling Language) figure of the overall architecture of Birdbuilder. |
|
Thomas Hamelryck* and Morten Kjeldgaard+
* ULTR department, Free University of Brussels (VUB), Paardenstraat 65,
B-1640 St-Genesius-Rode, Belgium.
+IMSB, Aarhus University, Gustav Wieds Vej 10C, 8000 Aarhus C, Denmark.
| Abstract |
| A computational framework for macromolecular crystallography is presented, consisting of a collection of freely available classes that perform various computational tasks, currently mainly including building and analyzing macromolecular crystal structures. These classes can be readily used or extended to quickly create simple applications, but they can also be used to build more complicated and elaborate programs. As an example of this, we present a program to interactively build, visualize and analyze macromolecular crystal structures. The project is currently in a preliminary state, and this short overview also serves as an invitation to join in the development of this Open Source project. |
The rapid growth of the amount of crystal structures of macromolecules puts high demands on the computational tools that scientists use to build, refine and analyze crystal structures. Traditionally, the crystallographic community has used a myriad of different, mostly FORTRAN, stand alone utilities, tied together in various improvised ways. In the future, it will be more and more difficult to use this approach, since the sheer number of structures will make a more integrated approach necessary.
Another, less benign evolution is the increasing spread of proprietary (and often expensive) scientific software, which cannot be modified, extended and/or properly understood. In addition, the procedures implemented in such programs are commonly not well documented, let alone published. This development is particularly surprising since the Open Source software development model is gaining more and more popularity in areas outside science.
Another impediment to future software development in the field of macromolecular crystallography is the declining number of students knowledgeable about programming, and the FORTRAN programming language in particular. It can therefore be foreseen that the maintenance and development of the many existing FORTRAN programs will be increasingly difficult. Add the fact that the basic algorithms in these programs tend to be obscured by the constructs required by the limitations of the language itself.
In this article we present a comprehensive software toolkit (called "Birdwash"), consisting of a set of classes that deal with various aspects of structure building, refinement and analysis. These classes can be used to rapidly create programs that deal with various computational problems. A very important aspect of the toolkit is that it is easy to extend with new classes, add novel features to existing classes or even integrate external software components, including external databases.
The toolkit encourages code reuse and distribution, and is subject to the GNU Public License (GPL).
The toolkit is implemented in Python, an interpreted, object oriented programming language. Python is easy to learn, freely available and has a rapidly growing user base. The main disadvantage of Python is that it is considerably slower than a compiled language like FORTRAN, C or C++. However, this problem is easily overcome by migrating speed critical parts of the program to extension modules written in a compiled language (typically C, C++ or even FORTRAN). For this reason, most of the time-demanding calculations will take place in binary program segments, and the interpreted Python language will merely control the flow of the procedure. The use of Python in scientific computing is rapidly catching on, and is used in related fields such as molecular modeling and visualization ( MMTK by Konrad Hinsen, PyMol by Warren DeLano and MSV by Michel Sanner), and crystal structure refinement (the PHENIX project by Paul Adams, Ralf Grosse-Kunstleve et al. of the Computational Crystallography Initiative). The Python programming language is also at the heart of the Nonius Collect program suite by R.W.W. Hooft, for the control data collection, integration of images, and image analysis.
One immediate advantage of the approach is the existence of a great number of available modules available for the Python language. The most important example is the Numerical Python (``Numpy'') module, which very efficiently implements vector and matrix operations, along with a number of associated procedures from the LAPACK library, such as matrix diagonalization. Lengthy numerical calculations are slow when performed extensively in Python itself. It is therefore desirable to perform as many calculations as possible in Numpy. Fortunately, it is often possible to recast existing algorithms in a set of array operations. Fig. 1 shows how a simple peak search algorithm can be formulated as a series of shift-compare operations on the entire electron-density array, using the rich set of array manipulating functions available in Numpy.
Another example of the use of standard Python extension modules, is the very efficient XML1 parser, which is used to parse the content of configuration and data files. This enables the storage of information about connectivity, bond angles, bond lengths, etc. in XML format, and allows for easy parsing, browsing and validation.
| Figure 1: A simple peaksearch algorithm implemented in Python, using the Numerical Python extension module. From the electron density Numpy array, different copies are made, each with a different shift of the map. These copies in fact share the same physical memory, but represent different modes of access. The ``greater'' function returns an array of elements with the value 1 if the condition is true, else 0. The ``nonzero'' function returns a list of indices where the array elements are different from zero. At the end of the computation, the ``peaklist'' array contains peak coordinates in grid space. The calculations takes around 5 seconds on a 400 MHz Pentium II processor. |
shift_zero = ed[1:-1, 1:-1, 1:-1] shift_left = ed[:-2, 1:-1, 1:-1] shift_right = ed[2:, 1:-1, 1:-1] shift_up = ed[1:-1, :-2, 1:-1] shift_down = ed[1:-1, 2:, 1:-1] shift_hither = ed[1:-1, 1:-1, :-2] shift_yon = ed[1:-1, 1:-1, 2:] flag = greater (shift_zero, shift_left) flag = flag * greater (shift_zero, shift_right) flag = flag * greater (shift_zero, shift_up) flag = flag * greater (shift_zero, shift_down) flag = flag * greater (shift_zero, shift_hither) flag = flag * greater (shift_zero, shift_yon) peaklist = nonzero(flag) |
Birdwash uses a structure/model/chain/residue/atom (SMCRA2) hierarchy to represent a crystal structure. Biopolymers like proteins or nucleic acids are grafted on this basic representation. New polymer types can easily be added by providing a simple description file in XML format that described bonds, angles and torsion angles in the polymer.
The SMCRA hierarchy is general enough to accommodate any type of biological polymer, as well as smaller entities such as waters, cofactors, etc. The classes used to represent a structure provide various operations like copy, delete, replace and add operations. However, while the SMCRA hierarchy contains all relevant information that can be perused from a PDB entry, it is not suited for computational tasks. The Structure class is typically handed to a computational client who extracts the information it needs. Symmetry expansion, for example, is handled by extracting all atomic positions into a Numpy array, which is multiplied with the symmetry operator and the atomic positions are reinserted into the SMCRA structure. While such an approach may seem cumbersome, it is extremely fast and strictly maintains the modular and cooperative approach of the toolkit.
Support for alternate atomic positions are fully supported by the toolkit, and from the point-of-view of the toolkit, there is no difference between an atom in one position, or one split between alternate sites. For example, it is possible to apply a rotamer transformation to the alternate configuration of a side chain. Birdwash also deals with anisotropic B factors if they are present.
To demonstrate a simple use of this Fig. 2 shows a Python code snippet downloads entry 1ABC from an FTP server and prints the average B factor of all C-alpha atoms. Afterwards the structure is deleted.
| Figure 2: Simple program using the toolkit. First, the program opens a parser with input from an FTP site. The (structure, model, chain, residue) hierarchy is traversed to extract the temperature factor of all C-alpha atoms, and the average temperature factor of these atoms is computed. |
from Birdwash.Parsers import PDBFTPParser
parser = PDBFTPParser("ftp.ebi.ac.uk", "/pub/pdb/")
structure = parser.get("pdb1abc.ent")
avb = 0
nr_atoms = 0
for model in structure.get_list():
for chain in model.get_list():
for residue in chain.get_list():
if residue.has_key["CA"]:
avb = av_b + residue["CA"].get_bfactor()
nr_atoms = nr_atoms+1
print "Average B factor of CA atoms", av_b/nr_atoms
|
The SMCRA hierarchy is a convenient way to store the information in a coordinate entry. However, this representation is in most cases too simple. For example, it contains no information about which atoms are bonded to which, and which residues are attached to which. This is the role of the Polymer classes. For example, from the XML residue dictionary, the Polymer classes knows about intra- and inter-residue restraints, and can therefore participate in structure regularization. In practice, a specific class exists to deal with a special polymer type. Most biological polymers are linear, so protein and RNA polymers are handled by classes which are subclasses of the LinearPolymer class, which in turn is a special subclass of the Polymer class.
Another higher level organization of molecular structures are that of biological assemblies of multimers. In crystal structures, such multimers may exist in the asymmetric unit of the crystal, or may be formed by the application of crystal symmetry to the asymmetric unit. A class hierarchy capable of handling biological entities in this manner will be developed in the future.
Birdwash contains parser classes for the PDB and the mmCIF format. Other file formats can be added easily since the code that deals with the actual building of the structure is stored in a separate builder class, and can thus be easily reused. Parsers are available that directly download structures from FTP repositories (see Fig. 2). The PDB parser is completely implemented in Python, whereas the mmCIF parser on the lowest level makes use of a lexical token analyzer written in C and lex. A parser capable of loading coordinate data directly from an SQL relational database is currently under development.
The Zope Object DataBase (ZODB) is a high-performance, transaction based database written in Python, that provides objects with Persistence storage and management. Persistence of the living objects in Birdwash is achieved using a ZODB database . This is especially important for the graphics program Birdbuilder.
Classes to deal with crystallographic symmetry are fully implemented in Birdwash. For example, methods are present to lookup symmetry operations based on space group name, to find crystal contacts and to apply arbitrary transformations to atomic co-ordinates. Internally, spacegroup lookup is implemented using Ralph Grosse-Kunstleve's SGInfo library .
Currently, only parts of the low level infrastructure dealing with structure factors have been developed. Structure factor file parsers exist for MTZ, CNS and mmCIF format. The mmCIF parser makes use of the same general mmCIF parser module being used for parsing coordinates, while the MTZ format is handled entirely in Python. On the lowest level, a Python module emulating the low level file i/o of the CCP4 suite is reading the binary data from disk. The actual data the parsers is being delivered in Numpy arrays. We are currently tracking the development of the Clipper library by Kevin Cowtan. Such an approach is very easily integrated into Birdwash, and fits well with the overall scheme.
The object oriented approach is attractive within crystallographic computing because it is straightforward and logical to deal with data as objects. For example, when thinking about a Fourier transformation of structure factors it is evident that applying an FFT operation on the structure factor object would return a map object (Fig. 3). The actual FFT is based on the Python bindings for the FFTW package which is under the GPL, but it may be advantageous to wrap Ten Eycks FFTLIB library, since it is fast and deals with crystallographic symmetry.
| Figure 3: Pseudo-code snippet demonstrating how crystallographic data items can conveniently be treated in an Object Oriented Programming approach. A reflection file is read and stored in the object ``data''. The appropriate data is extracted in columns and passed to the FFT procedure which generates a map object. |
data = hklFile("data.mtz")
fo = data["fobs"]
fc = data["fcalc"]
phi = data["phi"]
map = fft(2*fo-fc, phi)
map.write("map.cdf")
|
In Birdwash, structure factors are stored in netCDF (network Common Data Form) files. The netCDF format is actively being developed by the Unidata Program Center in Boulder, Colorado, and defines a machine-independent, self-describing format for representing scientific data. Multidimensional data may be accessed one point at a time, in cross-sections, or all at once. Data is directly accessible, permitting efficient access to small subsets of large data sets. Bindings for various programming languages such as C, C++, FORTRAN, Perl or Java exist, and Python bindings that deliver the data in form of Numpy arrays have been developed by Konrad Hinsen. Utilities for converting structure factor information to and from netCDF data format exist as a part of Birdwash and are easy to program. We encourage the crystallographic community to consider the netCDF format as a truly portable, cross-platform general purpose format for binary data.
The low level infrastructure for dealing with electron density maps is available in the toolkit. Currently, data from CCP4 and CNS format density maps can be imported. The three-dimensional contouring in Birdbuilder (see section 5) is carried out by a Python module which in turn calls a two-dimensional contouring routine implemented in C. Remarkably, there is no noticeable speed penalty in this implementation compared to the equivalent functionality in O [1].
The Birdwash generic persistence format for electron density maps (and masks) is again based on the netCDF standard, offering platform independence and random access.
Birdbuilder consists of three large parts: the structure manager, the command manager and the GUI (Fig. 4).
| Figure 4: UML (Universal Modeling Language) figure of the overall architecture of Birdbuilder. |
|
|
Every action performed on a loaded structure is performed by calling the execute method of a specific Command class. These Command classes are loaded on program startup by the command manager. The command manager also takes care of undo operations by calling the undo methods of the Command classes involved. This architecture allow the moderately advanced user to add novel commands to the program, simply by writing a new Command class and adhering to a couple of conventions. If the user supplies an ``Undo'' method in a Command class, the actions are reversible as well.
Note that Birdbuilder does not simply provide a macro facility: it provides a full blown programming environment that can be used to extend the program. It also allows the user to install only the commands that are relevant to the intended use of Birdbuilder (eg. it is not necessary to install commands that deal with nucleic acids if the program will only work with proteins).
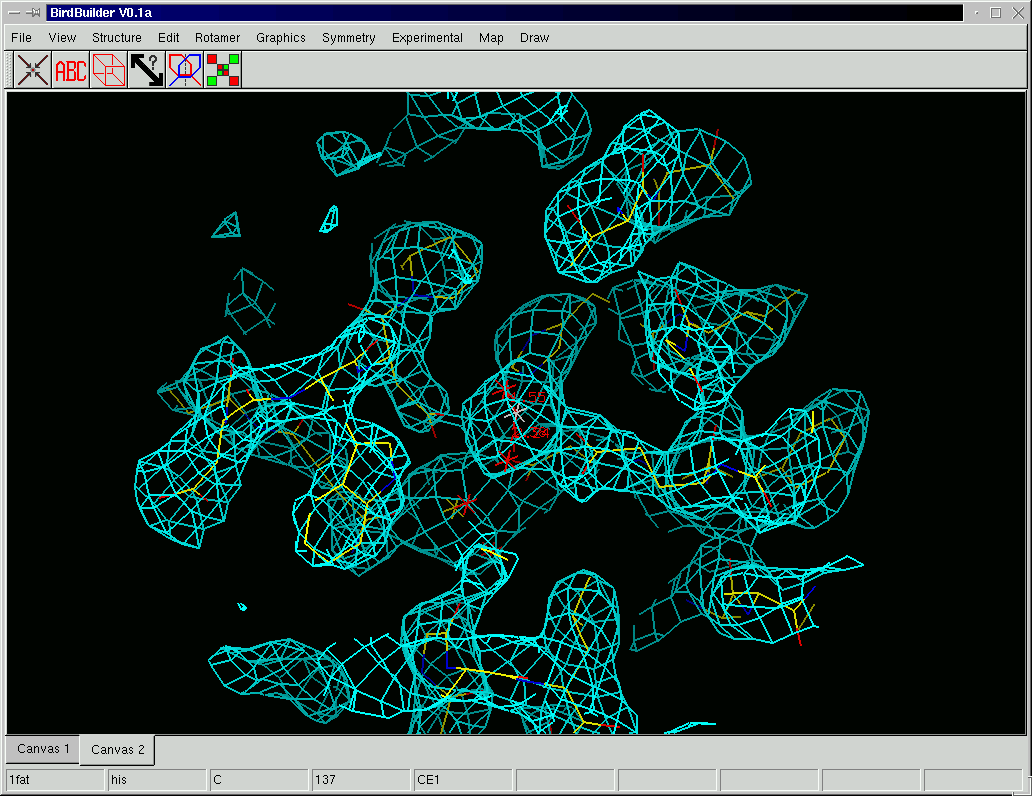
Birdbuilder (see Fig. 5 for a screenshot) currently incorporates a number of the features expected in an interactive molecular graphics program. The current version although still in early development contains (unordered list):
|
| Figure 5: A screenshot of Birdbuilder with a contoured electron-density map. |
An important aspect of Birdbuilder will be interaction with various databases. Currently, the interaction with many databases typically occurs through a web based interface. This is fine for casual users, but more experienced users would like to see a more sophisticated interface to these databases. We are currently trying to put our efforts in tune with external database efforts. Birdbuilder has the necessary tools to efficiently interact with external databases over the Internet, either using simple SQL queries or via a more advanced CORBA interface.
As a first start, we have implemented the automatic retrieval of a structure from the PDB database (ie. you provide a PDB identifier and the structure immediately pops up on the screen). More advanced database connectivity will need the co-operation of the various maintainers of these databases.
The current project started in late 1999, and has been under development for a little over a year. We were initially curious about the idea of programming a major program system like a molecular graphics program in an interpreted language like Python. Indeed, it appears to be a crazy idea! It was not at all clear at the time that the speed of such a program be acceptable. Time and experience has shown that the choice was right. From working with the system, and developing it, we have experienced that the speed of development is probably a factor of 3-4 faster than using a compiled language. This is also partly due to the advantages of using an Object Oriented programming method, partly to due to the extremely rich selection of ``add-on'' Python modules on the Internet, and partly due to the rich set of build-in types in Python, such as lists and dictionaries. With these tools, one can very easily test algorithms and implementations without constantly having to hand-craft these basic data structures.
The Numerical Python module is an essential prerequisite of any serious computation using Python. It is extremely efficient, versatile, and fun to work with. And, last but not least, is actively developed and maintained.
There was never a doubt that the three-dimensional graphics API for Birdbuilder was going to be OpenGL. Finding the right GUI took a bit of testing, until we finally settled on wxPython, which we now realize was the best choice. It is very complete, robust, and flexible, and again, actively and enthusiastically maintained.
A main design decision was to make use of existing libraries where available on the Internet. Why spend time programming a conjugate gradient or simulated annealing refinement procedure, when it has already been done by specialists?
Birdbuilder and the Birdwash toolkit are still in an early experimental stage. At this point, we have constructed the basic data structures and a basic functionality, and we are hoping to gradually involve other daring computer-nerds and programmers in the project. It is still a long way from being a tool for the Casual User. However, it is our experience that many students of crystallography in their projects meet problems that require some programming to be done. Rather than starting from scratch, and having to work out a bunch of basic problems that have already been solved, it is better to stand on the shoulder of others, and spend more time on the problem itself.
All software components can in principle run on many different platforms, including such exotic ones as MS Windows. However, the hardware mainly targeted is an mid-upper range PC running Linux. We supply a set of RPM packages for the extra software needed to run the package. The Birdbuilder/Birdwash directory tree is currently only available as a gzipped tar archive.
Additional information is available on the Birdwash home page.
TWH has a grant from the Fonds voor Wetenschappelijk Onderzoek (FWO). MK is the recipient of a Hallas-Mřller fellowship from the Novo-Nordisk Foundation.
[1] T. A. Jones and J-Y. Zou and S. W. Cowan and M. Kjeldgaard (1991) ``Improved Methods for Building Protein Models in Electron Density Maps and the Location of Errors in these Models'', Acta Cryst. A47, 110-119.
1eXtensible Markup Language
2pronounced: simcra