

------------ CCP4 Newsletter - June 1996 ------------
IGBMC du CNRS,
Parc d'Innovation,
BP 163,
67404 Illkirch,
c.u. de Strasbourg, France
IMPB of RAS,
Puschino,
Moscow Region,
142292, Russia
The final purpose of the molecular replacement method (Rossmann & Blow, 1962; Crowther & Blow, 1967; Rossmann, 1990; Rossmann & Arnold, 1993; and references therein) is to find the position of a given search model in a given crystal. Another meaning of the term "molecular replacement", as a procedure to improve density distribution by averaging through non-crystallographic symmetry, is also widely used, however, to avoid any misleading, is not considered further in these notes. The search of the internal crystal symmetry (self-rotation analysis) as a tool to improve a molecular image is not considered here either.
A search atomic model is defined by a set of three-dimensional coordinates given in some absolute or external Cartesian coordinate system. The result of the molecular replacement search is a transformation which should be applied to these coordinates. The transformation can always be presented as a rotation plus a translation.
There is a number of different descriptions of rotations in three-dimensional space reflecting point of view of different authors. These variants differ by the definition of :
The program CONVROT is developed which calculates the rotation matrix for different types of rotation description and vice versa. Such approach solves two problems. First, it gives directly a rotation matrix which can be applied to the model coordinates in order to obtain a model orientation accordingly to the molecular replacement solution. Secondly, it allows to recalculate the rotational angles from any to any of these systems by use of 2N subroutines and not N*N, where N is the number of different rotation systems : N subroutines to go from rotation angles to the rotation matrix and N - for opposite operations. This also allows easy addition of any new rotation system.
The program can define all symmetry related rotation angles for a given one when the orthogonalisation agreement and the symmetry operations are known.
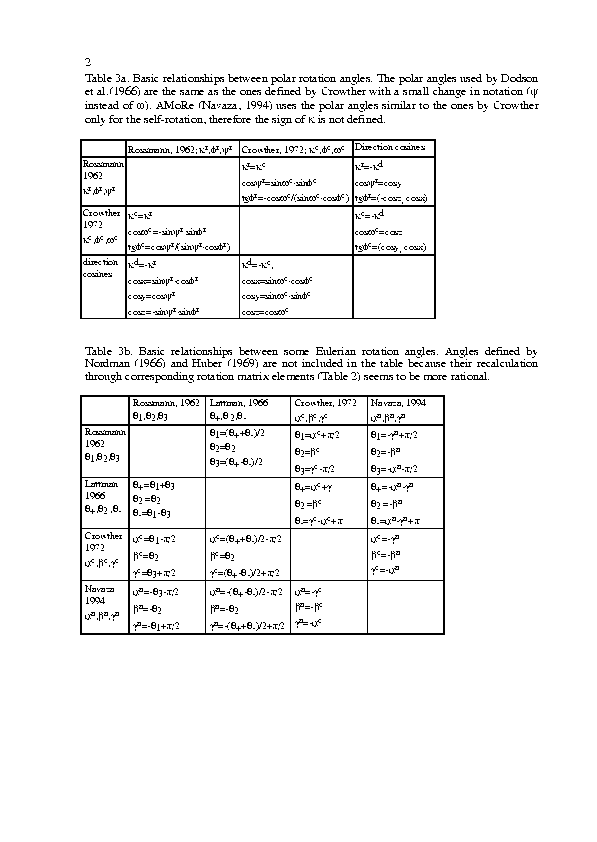
The program can work with the polar systems introduced by Rossmann & Blow (1962), Dodson et al. (1966), Crowther (1972, 1973) and the eulerian angles systems introduced by Rossmann & Blow (1962), Nordman (1966), Huber (1969), Crowther (1972, 1973), Lattman (1972), Navaza (1994). The system of direction cosines is also used (see, for example, Diamond, 1993). These system are used, in particular, in ALMN (Crowther, 1973; CCP4, 1994), in PROTEIN (Steigemann, 1974), in MERLOT (Fitzgerald, 1988), in X-PLOR (Brünger, 1992) and in AMoRe (Navaza, 1994).
Tables 1-2 give some basic formulae used by the program, the singular cases are not included. [Click on tables to obtain postscript version.] Table 1 uses the following notation:
| cosx -sinx 0 | RZ(x) = | sinx cosx 0 | | 0 0 1 | | cosx 0 sinx | RY(x) = | 0 1 0 | | -sinx 0 cosx | | 1 0 0 | RX(x) = | 0 cosx -sinx | | 0 sinx cosx |
Table 3 gives some simplest relationships between rotation angles.
The program is written on standard FORTRAN-77. The source is available by request from the author. Corresponding e-mail address is sacha@igbmc.u- strasbg.fr. Any reported comments, found mistakes and formulae improvements are appreciated. The full text of the notes will be published elsewhere. A new user-friendly version of the program is under development.
The author thanks Drs. A.Poterszman and J.C.Thierry for pointing the problem which initiated this work, Drs. J.Navaza, A.Podjarny and B.Rees for their extremely useful discussions about molecular replacement and Dr. D.Moras for his interest to the work. The work was supported by the CNRS through the UPR 9004, by the Institut National de la Santé et de la Recherche Médicale and the Centre Hospitalier Universitaire Régional.
One disagreement was indicated by Ulrich Baumann : the PROTEIN rotation matrices are inverted in comparison with the X-PLOR ones and do not coincide with them. Therefore, in Table 1 corresponding matrices for PROTEIN are :
Polar :
Ry(phi) Rz(-psi) Ry(kappa) Rz(psi) Ry(-phi)
(excluded from line 1 of Table 1)
Eulerian :
Rz(theta1) Rx(theta2) Rz(theta3)
(not shown in Table 1; PROTEIN can use this Eulerian
system as an alternative to the one by Huber)
The corrected Tables are included in the current version of this article due to kind permission of the CCP4 staff.
The full version of this material is in preparation and is supposed to be sent to J.Appl.Cryst.