|
CCP4 Molecular Graphics Documentation |
 |
| Atom Selection |
Introduction
An atom selection menu is opened from the Display Table by clicking the Atom selection button for a variety of model display objects.
 The options on the atom selection menu are customised for the data; for example
the chains and monomers in the structure will be listed where appropriate.
Several options on the selection menu have pull-right sub-menus for
selecting individual items (for example for Monomers).
The options on the atom selection menu are customised for the data; for example
the chains and monomers in the structure will be listed where appropriate.
Several options on the selection menu have pull-right sub-menus for
selecting individual items (for example for Monomers).
The Restore selection option opens a window which lists any saved selections. The saved selections are listed in two groups; either for the particular model or for All models. How to save selections is described later.
The Selection browser option opens the Selection Browser..
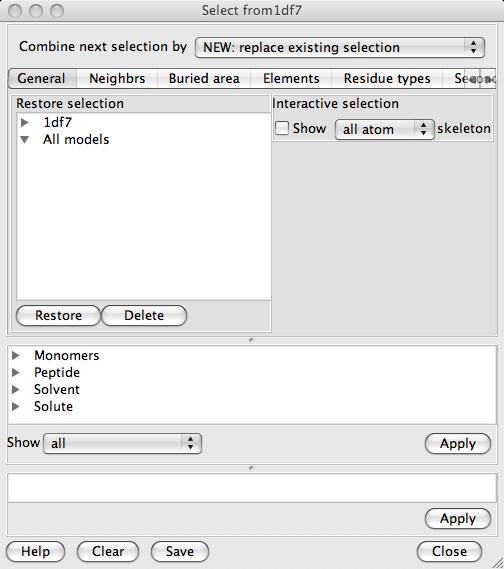 The browser consists of three vertical sections with three different approaches to atom selection:
The browser consists of three vertical sections with three different approaches to atom selection:
The Tab frame has pages with specialised tools such as 'Neighbours' and 'Secondary structure'.
The Selection tree is a tree view of model with the main branches being the chemical type such as 'solvent', 'peptide' or 'monomer' and the successive branches being chains, residues and atoms.
The Command text section shows the text command. The text command is generated automatically from the input to the other sections of the window and is sent to the program selection interpreter. The command text can be edited manually by 'expert' users.
The three sections are separated by pane dividers which can be dragged up and down to expand and contract the different sections. Note that the tab frame is a fixed size so will be either open or closed. Each section of the interface has an Apply button which will apply the current settings in that section and update the graphical display. The Apply button in a tab tool updates both the tree view and the command text. The tree view Apply updates the command text and the command text Apply updates the tree view.
The Tab Frame
Combine next selection
At the top of the tab frame is the combine next selection combo-box to control how any newly applied selections are combined with any pre-existing selection. The default NEW: replace existing selection will completely override any pre-existing selection. The combine next selection setting applies to all tab pages except the Restore selection (which always overrides any pre-existing selection) and it also applies to interactive selections. Beware that in everyday speech the word 'and' is ambiguous: when you are saying 'and' you may want the logical operator 'OR'.
General tools and interactive selection
The General page has several sections. On the left side is a list of saved selections which can be restored by double clicking on the appropriate line or by selecting the line and clicking the Restore button. Selecting a line and clicking the delete button will remove the saved selection. The current selection can be saved by clicking the Save button at the bottom of the window and entering a short name for the selection. Note that the information for saved selections is stored in the model definition file.
While the selection browser window is open it is possible to select atoms, side chains, main chains, residues, residue ranges, secondary structures or chains from the graphical display. Clicking on an atom with the right mouse button opens a context menu with a Select (new) this item which has a sub-menu to choose atom, residue, start residue range etc.. The interactively selected atom(s) will combined with any pre-existing selection according to the current status of the combine next selection menu. For you to to be able to pick atoms in interactive selection the atoms must already be displayed so there is an option to display a skeleton model (see the right side of the General page).
Dependent on the composition of the model the General page may also have check buttons to control display of alternate location atoms and/or hydrogen atoms.
Neighbours
The Neighbours page enables selection of atoms within a given distance of a central group of atoms. This is effective for displaying the binding site of ligands.
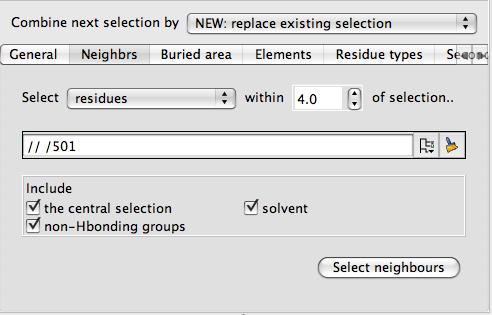
All atoms in a group (such as residue or side chain) which has any atoms within the cutoff distance of the central atom(s) will be selected. Note that the group option 'main/side chains' usually gives a good representation of a binding site by treating each residue as two groups of atoms and selecting a group only if it has a atom within the cutoff distance. The central group of atoms are selected using the standard atom selection combo (see tutorial on Find/CentreOn). You can choose whether to include the central atoms or solvent in the selection or can limit the selection to groups that hydrogen bond to the central atoms. Click Select neighbours to apply the selection.
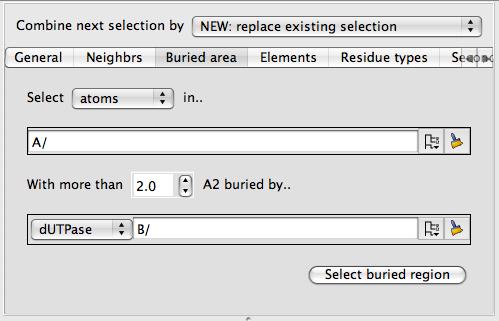
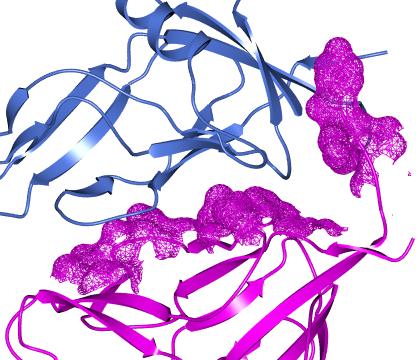
Buried area
This tool will select those atoms or residues on the surface of one set of atoms that are 'buried' (i.e. interfacing to) another set of atoms. This tool is particularly useful applied to surfaces.
As in the example above you should select two groups of atoms (such as two different chains) and click Select buried region. Beware that this tool may be computationally expensive. If you are applying this tool to a surface then you will also need to select the context set of atoms (the second column in the display table)

which will usually be the same as the first set of atoms in the buried area tool.
Elements and residue types
These two tools have similar interfaces with a list of the elements or residue types on the left side of the interface.
Secondary structure elements
The secondary structure elements are listed on the left side of the tools, and there are buttons to select either all beta strands or all alpha helices.
Properties
This tool enables selection by temperature factor, occupancy, atom coordinate, atom or residue solvent accessible area, atom or residue buried area or atom serial number.
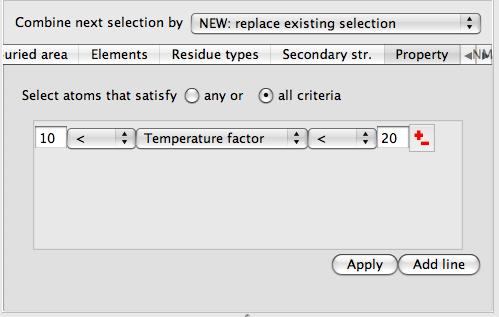
On opening the tool you will need to click the Add line button at the bottom of the tool. In the middle of each line is a menu to choose the property that is being selected for. You should type in the lower and upper limit for the property value. Note also the choice of selecting atoms that obey all or obey any of the criteria.
NMR/symmetry models
Atom coordinate files from an NMR structure solution contain multiple models. The models have identical composition but different atomic coordinates representing different possible solutions. The models are identified numerically 1 to n where n is the number of models. CCP4mg also uses the 'NMR' models to hold the coordinates for symmetry related copies of the original atoms; see the generate symmetry mates documentation for more information. In the CCP4 selection language the model is the top level of the hierarchy of model, chain, residue and atom. This selection tool enables selection of NMR or symmetry models. The interface list the models with the model number and the symmetry id for symmetry models.
The Selection Tree
The middle section of the selection browser contains a tree representation of the model with the main branches of the tree being the chemical components such as peptide, solvent, monomers etc. and the sub-branches being chains, residues and atoms. Note that the NMR/symmetry models are not represented in the tree. Items at any level in the hierarchy can be selected and, beware, if a parent item is selected then all of its children are selected. The tree selection is only applied after clicking the Apply button. The tree selection of chains and/or residue ranges are combined with selections such as 'CA trace','main chain' in the combo-box beneath the tree widget. By default this combo-box is set to 'all' which has no effect but any other selection will be AND'ed with the tree selection.
The tree selection is updated when a tab tool, an interactive selection or the command text are applied.
The Command Text
The bottom section of the selection browser is a text input box which is automatically updated to show the selection command derived from other user interface input. Entering commands directly to this box can sometimes be quicker particularly for small corrections. Editing the command text is also more powerful for generating complex selection commands.
Save current selection
The Save button at the bottom of the selection browser window will save the current selection command with the option for it to be accessible for
- The present model: the selection is saved to a file called model_name.ccp4mg and is only available to use with present model.
- All models: the selection is saved to a file called $HOME/.CCP4MG/generic.ccp4mg and is available for all models.
You should enter a short one-word name for the selection.
The Selection Widget
This widget appears in many interfaces within CCP4mg. The widget is customised according to the context.

The widget consists of:
a text entry box in which you can enter a selection command
 the tree view icon to open the tree view of the model
the tree view icon to open the tree view of the model
 the clear icon which clears selections
the clear icon which clears selections
 the lasso icon - click on this to popup a menu of selection tools
the lasso icon - click on this to popup a menu of selection tools
The tree view which is opened by clicking on the icon works like a
file browser with the model split at the first level into peptide, monomers and
solvent etc.. Clicking on arrow beside the folder title will open the folder. Clicking on items in the tree will select them. The selections made in the tree view will be shown in the text entry box. Beware that if a folder title line is selected then everything in the folder will be selected. The tree view can be closed by clicking elsewhere in
the window or hitting the Esc button.
The icon is not always present and the items on the menu will be appropriate for the context. Choosing a selection tool from the menu will open a new window with one of the tools described above in this page. When the selection is applied the selection command is shown in the text entry box.
Selection Commands
A selection command is a text string which defines a set of selected atoms. All user interface input is converted to a selection command and the command is interpreted to determine which atoms are displayed. By editing the selection command directly you can set up more complex selections than are accessible via the user interface. The selection command can be viewed or edited using the Enter selection or Interactive selection - Advanced tools.
The components of selection commands are
- The coordinate id (CID) specifies a particular atom, residue or chain and can also specify a range of residues, residue types, group of atoms etc.. For example 'A/27/CA' is the CID for the CA atom of residue 27 in chain A.
- Aliases are one word aliases for more complex CIDs. For example the alias 'catrace' will select all atoms called 'CA' in amino acid residues.
- A Command is a keyword followed by a variable number of arguments. For example 'neighb cid="A/27" maxd=5.0' will select all atoms in the neighbourhood, that is within 5 Angstrom, of residue 27 of chain A.
- The Operators combine CIDs, aliases and commands to create more complex selections. For example the rule '(GLU,ASP)' and 'neighb cid="A/27" maxd=20' combines the CID '(GLU,ASP)' which selects residue types GLU or ASP and the command 'neighb cid="A/27" maxd=20' which will select all residues within 20A of A/27. Combining these components with the 'and' operator will select atoms in GLU or ASP residues within 20A of A/27
Coordinate IDs
A coordinate ID is an text string that has the following format:
/mdl/chn/seq(res).ic/atm[elm]:aloc
where
| Item |
Description |
| mdl |
the number of the model (usually an NMR model) |
| chn |
chain ID |
| seq |
residue sequence number |
| res |
residue name |
| ic |
residue insertion code |
| atm |
atom name |
| elm |
chemical element ID |
| aloc |
alternative location indicator |
The residue name and atom element type are usually redundant, that is they are not essential to uniquely identify an atom, and do not need to be included when entering a CID. But note that all spaces in the coordinate ID are ignored and this means that certain
atom names will be interpreted correctly only if the chemical element
name is supplied (compare Calcium CA[CA] and
Carbon in alpha-position CA[C]).
Any item in the coordinate ID may be replaced by a wildcard
"*", which means an indefinite value for
that item. The wildcard value is automatically implied for any
missing item except the chain ID, insertion code and alternative
location indicator:
- A missing chain ID is interpreted as an empty ID
"" if there is a slash "/"
after it. In all other cases, the missing chain ID is
interpreted as a wildcard "*".
- A missing insertion code is interpreted as a wildcard
"*" if the residue sequence number is
not specified or set to a wildcard. In all other cases,
a missing insertion code is interpreted as
"" ('no insertion code').
- A missing alternative location indicator is defaulted
to a wildcard "*" if no atom name
or chemical element name is given. If any value for
atom and/or chemical element name is given , even though a
wildcard, then a missing alternative location
indicator is defaulted to ""
('no alternative location indicator').
A coordinate ID may be incomplete. Below are the rules for
interpretation of incomplete IDs. Curling brackets
{..} denote parts of an ID string that may
be omitted as a whole:
- If the ID starts with slash "/" then
it is interpreted as
/mdl { /chn { /seq(res).ic { /atm[elm]:aloc }}}
- If the ID starts with a letter:
chn { /seq(res).ic { /atm[elm]:aloc }}
- If ID starts with a (possibly negative) number,
bracket "(" or dot
".":
seq(res).ic { /atm[elm]:aloc }
- If none of the previous cases apply but the ID string contains
square bracket "[" or colon
":", it is interpreted as
atm[elm]:aloc
Below are examples of valid coordinate IDs:
| Coordinate ID |
Description |
| /1/A/33(SER).B/CA[C].A |
model 1, chain A, residue SER with sequence
number 33 and insertion code B, C-alpha atom in alternative
location A. |
| /1/A/*(SER).* |
any SER residue in chain A, model 1.
|
| /1/A/(SER) |
any SER residue with no insertion code in chain A, model 1. |
| /1//(SER) |
any SER residue with no insertion code in chain without a chain ID, model 1. |
| /1/A/*.*/CA[C] |
any C-alpha atom with no alternative location indicator in chain A, model 1, in residues with any sequence number and insertion code. |
| /1/A/*/CA[C] |
any C-alpha atom with no alternative location indicator in chain A, model 1, in residues with any sequence number and no insertion code. |
| A/*/CA |
any C-alpha or Calcium atom with no alternative location indicator in chain A of any model, in residues with any sequence number and no insertion code. |
| CA[C] |
any C-alpha atom with no alternative location indicator in any chain, any model, in residues with any sequence number and no insertion code. |
| CA |
any atom of chain CA with no alternative location indicator, in any model, in residues with any sequence number and no insertion code. |
The following coordinate IDs are incorrect:
1. /A/23/CA[C] stating with a slash implies that the model number will be given
2. /1/CG[C] the residue is not defined
3. /-15 stating with a slash implies that the model number will be given
4. */*(*).*/*[*]:*
Selection Aliases
Selection aliases are one word aliases for complex CIDs. The predefined selection aliases are specified in the file ccp4mg/python/ui/selection_protocols.py. This needs a nice GUI to allow customisation. Within the program the implementation of selection aliases is different from selection commands but the user need not be concerned by this.
| amino_acid |
All amino acid residues - defined as a list of residue types |
| nucleic |
All nucleic acid residues - defined as a list of residue types |
| solvent |
All solvent - defined as a list of residue types |
| solute |
All solute - defined as a list of residue types |
| catrace |
All atoms called 'CA' in amino acid residues |
| peptide_link |
The main chain atoms N,C,O,H |
| main |
All main chain atoms in amino acid residues |
| side |
All not main chain atoms in amino acid residues |
| ca+side |
All CA and side chain atoms in amino acid residues |
| backbone |
Atoms C3*,O3*,H3*,C4*,C5*,1H5*,O5*,2H5*,H5T,P,O1P,O2P |
| sugar_ring |
Atoms C1*,H1*,C2*,2H2*,1H2*,C3*,O3*,H3*,C4*,H4*,C5*,1H5*,2H5*,O5*,H5T,O4*,O2* |
| base |
Defined as the atoms which are not backbone or sugar_ring |
| polar_atoms |
Defined as the atoms of element type N,O,P |
Selection Commands
More complex selections can be defined by a selection command which consists of a command keyword and a variable number of arguments.
The format for commands is:
command [arg1 [arg2 .... [argn]]]
with the arguments in the order given in the descriptions below.
Currently supported commands and their arguments:
| neighb |
Select atoms, residues or chains within neighbourhood of given object |
| cid |
An ID for the central atom, residue or chain |
| maxd |
The maximum distance from the central atom to the neighbours |
| group |
Select all atoms in a group that have at least one atom within maxd distance. The currently supported groups are: model,chain,residue,main,side,catrace,main_side,CA+side,solvent_monomers. |
| hbonded |
If this has value 1 then only hydrogen bonded atoms are selected. |
| excl |
A comma separated list of atoms which will be excluded from the selection: central (the 'central' atoms defined by cid argument), solvent, monomer (all monomers), peptide (main chain atoms excluding CA). |
| sphere |
Select atoms, residues or chains within radius of a given x,y,z coordinate |
| x |
X coordinate |
| y |
Y coordinate |
| z |
Z coordinate |
| radius |
The maximum distance from the central point |
| type |
Should be 'atom','residue' or 'chain' to specify type of objects selected |
| all |
|
>Select all atoms in the model |
| oneatom |
|
>Select one atom per residue in the model |
| termini |
|
Select the termini residues of chains |
| symid |
Select the model containing coordinates for the symmetry mate |
| n_xyz |
symmetry id |
Selection Operators
Complex selection commands are built up from CIDs, aliases and commands connected by the operators
| a or b |
select all atoms defined by a and b |
| a and b |
select only the atoms in both a and b |
| not a |
select atoms that are not in a |
| a xor b |
select all atoms that are in a or b but not in both |
| a excl b |
select the atoms that are in a but exclude those in b. This is equivalent to 'and not'. |
The selection components can also be grouped by using curly brace. Note that curly brace are used to be distinct from the brackets used to denote residue types in the CID.
Examples of Selection Commands
amino_acid and not main
uses the aliases amino_acid and main to select atoms in amino acids that are not main chain (i.e. side chain)
(GLU,ASP)/(O) and neighb (HIS)/NE2,ND1 5
Select oxygen atoms in glutamic acid or aspartic acid residues within 5A of the ND1 or NE2 of a histidine residue
{ main and {A/10-25 or A/35-40} } or (MTX)
Select the main chain atoms for two given ranges of residues and any residue with residue name MTX