AMPLE – using ab initio modelling to tackle difficult molecular replacement cases
Jaclyn Bibby1, Ronan Keegan2, Olga Mayans1, Martyn Winn3, Daniel J. Rigden1
1Institute of Integrative Biology, University of Liverpool, Liverpool L69 7ZB, UK
2RCaH, STFC Rutherford Appleton Laboratory, Chilton OX110FA, UK
3STFC Daresbury Laboratory, Warrington WA44AD, UK
AMPLE is a joint software development by the University of Liverpool and CCP4. The project’s main aim is to assess the suitability of using cheaply obtained ab initio models as search models in molecular replacement. An additional goal of the project is to make AMPLE into an automated software pipeline tool that can be made available to CCP4 users. This new tool can be used to generate candidate search models for use in molecular replacement problems where success cannot be achieved using homologous structures from other sources or in cases where no obvious homologous structure is available.
Ab initio structure prediction is the prediction of a target structure fold based purely on its sequence information. Some examples of ab initio protein structure prediction software are ROSETTA (http://www.rosettacommons.org/) and QUARK (http://zhanglab.ccmb.med.umich.edu/QUARK/). The first step these programs typically perform is to generate thousands of what are known as “decoy” models. These are rough predictions of the target structure created by a fragment assembly process using libraries of fragments determined for overlapping ranges of the target. Fragments are selected from the PDB based on sequence similarity and consistency with predicted secondary structure. These decoys are clustered based on tertiary structure to give an idea of the most likely fold for the target. A large cluster of similarly folded decoys is indicative of a correct prediction. This first step is relatively quick (~1 minute of CPU time per model). The next step is to add side chains followed by the refinement of the most likely folds under more realistic physics-based force fields. This step can require significantly more computing power and can take up to 100 CPU days to complete. AMPLE’s approach is to use ROSETTA to perform the first of these steps (decoy assembly and clustering) and to derive from the “cheaply obtained” clustered decoys a set of suitable search models for use in molecular replacement. This method was shown to work in a significant fraction of test cases in a pilot project (see Rigden et al. 2008). The program is currently configured to work with ROSETTA but models can be input from other sources manually such as those generated using QUARK.

Figure 1: Cluster of decoy models
A spin-off benefit of using these clustered decoys to generate search models is that they naturally lend themselves to the production of “ensemble” search models. Ensembles have been shown to make excellent molecular replacement search models and on some occasions can give a correctly positioned solution where any of the constituent search models that make up the ensemble fail. The general procedure for the processing of decoys into search models is described in detail elsewhere (Acta Cryst. D, currently in submission) but the end result is a set of several hundred ensemble models derived from the initial clustered decoys. Several processes are applied to the clusters to give a broad range of ensembles including many degrees of truncation based on a variance and RMS difference score between the decoys and three different degrees of side chain addition (polyalanine, most reliable side chains and all side chains)
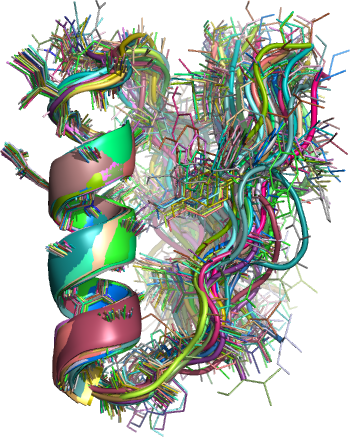
Figure 2: Example ensemble model
MrBUMP (Keegan and Winn, 2008) from the CCP4 suite is then used as the engine for processing all of these search models in molecular replacement. It automates the passing of the models to PHASER (McCoy et al., 2007) and MOLREP (Vagin and Teplyakov, 1997) and will refine the positioned models output from these programs using REFMAC5 (Murshudov et al., 1997) to give an initial estimate of whether or not they are a correct solution. An optional follow-on step, given sufficient resolution (2.2Å or better), of using the phase improvement and Cα-tracing options in the latest SHELXE (Sheldrick, 2008) can be performed to give a clear answer as to whether or not MR has been successful. A CC value for the partially traced structure against the native data of 25% or more is a solid indication of success and provides a suitable starting model for model completion.
A comprehensive test of AMPLE was carried out using a set of 295 structures from the PDB. The set was made up of structures with maximum sequence length of 120 residues and resolution of 2.2Å or better. Current limitations mean that it is less reliable for larger structures however it has been found to work for a structure as big as 240 residues. A mix of all-α, all-β, and mixed α – β secondary structures were selected. In the fragment generation step only fragments from non-homologous structures were used to generate decoys. Results showed that a correct solution could be found in 43% of cases. Success was heavily dependent on secondary structure type. For all-α targets, the success rate rose to 79% whereas all-β targets were difficult to solve and the success rate for these cases was just 3%.
A beta test release version of AMPLE is included in the latest release of the CCP4 software suite (version 6.3.0) with a ccp4i interface for ease of use. Several non-CCP4 packages are needed to make it work (including ROSETTA). Details of what dependencies are needed and how to run the program are available in the ccp4 wiki page for AMPLE (www.ccp4wiki.org).
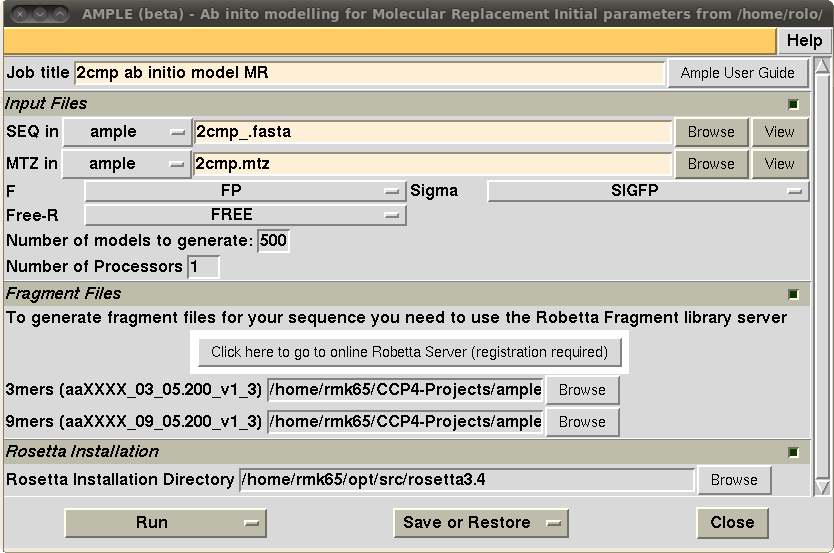
Figure 3: the ccp4i interface for AMPLE
References
Rigden, D.J., Keegan, R.M. and Winn, M.D. “Molecular replacement using ab initio polyalanine models generated with ROSETTA” (2008) Acta Cryst. D64, 1288-1291
Keegan, R.M. and Winn, M.D. “MrBUMP: an automated pipeline for molecular replacement” (2008) Acta Cryst. D64, 119-124
McCoy, A.J., Grosse-Kunstleve, R.W., Adams, P.D., Winn, M.D., Storoni, L.C., Read, R.J. “Phaser crystallographic software” (2007) J. Appl. Cryst. 40, 658-674
Vagin, A. and Teplyakov, A. “MOLREP: an automated program for molecular replacement” (1997) J. Appl. Cryst. 30, 1022-1025
Murshudov, G.N., Vagin, A.A. and Dodson, E.J. “Refinement of Macromolecular Structures by the Maximum-Likelihood Method” (1997) Acta Cryst. D53, 240-255
Sheldrick, G.M. "A short history
of SHELX" (2008).
Acta Cryst. A64, 112-122