Ian J. Tickle, Astex Therapeutics Ltd.
The CCP4 program AREAIMOL, originally written by Peter Brick (Imperial), for computing the solvent-accessible surface area from co-ordinates in PDB format (see Briggs, 2000 for more information), is frequently used to perform buried surface area calculations when a complex is formed. This requires taking the difference between two large numbers (i.e. the sum of the solvent-accessible surface areas of the separate molecules minus the surface area of the complex). The buried surface area may be only 5-10% of the accessible area so a small relative error in the accessible area can be multiplied 10-20 times. Hence it's important to make the area calculation as accurate as possible. AREAIMOL uses a surface point counting algorithm but it is critical that the distribution of points is as uniform as possible, i.e. the local surface point density is the same everywhere (note that this is not the same problem as randomly distributing points with a uniform probability distribution over the surface of a sphere, since then the local surface point density will have a statistical variation). Except for special values of the number of points on the surface of a sphere (corresponding to the numbers of vertices for the Platonic solids: 4, 6, 8, 12 & 20) an exact solution is not possible, and anyway for sufficient accuracy it's normally necessary to have 1500-2000 points per atom, so an approximate solution is needed. It was noticed that the algorithm used didn't perform very well in the polar regions of the sphere, particularly at low point densities, thus contributing to inaccuracies in the area calculation.
I performed some tests with two C atoms separated along the z axis by the sum of their VDW radii (3.6 Ĺ), varying the surface point density input to AREAIMOL and got the solvent-accessible surface area results with the distributed version shown in the Figure (labelled OLD). As can be seen the computed area varies significantly; an exact calculation using geometry, labelled CALC, gives 201.06 Ĺ2 ( = 4p (1.8+1.4)(1.8+1.4+3.6/2) where the VDW radius is 1.8 and the probe radius is 1.4). The distributed version of AREAIMOL uses a default surface point density of 1 Ĺ-2 which is clearly much too low for accurate calculations.
For reference here's the exact PDB file that was used for the surface area calculation for the 2-atom molecule:
CRYST1 57.023 64.684 188.747 90.00 90.00 90.00 P 21 21 21 ATOM 4 C LEU A 22 0 0 -1.8 1 0 ATOM 4 C LEU B 22 0 0 1.8 1 0
The errors arise because of a non-uniform distribution of points over the sphere: the problem is that the algorithm used distributes the points in circles of constant latitude but takes no account of the relative positions of points in adjacent circles. I looked up better algorithms and found a spiral distribution algorithm (Saff & Kuijlaars, 1997) which distributes a specified number of points in a spiral pattern over the surface of the sphere; it's a deceptively simple algorithm, the Fortran code (about half as many lines as the original point-generating code!) is:
DO I=1,N
CTHETA=2*REAL(I-1)/(N-1)-1
STHETA=SQRT(1-CTHETA**2)
IF (I.EQ.1 .OR. I.EQ.N) THEN
PHI=0
ELSE
PHI=PHI+3.6/(STHETA*SQRT(REAL(N)))
ENDIF
X(1,I)=R*STHETA*COS(PHI)
X(2,I)=R*STHETA*SIN(PHI)
X(3,I)=R*CTHETA
ENDDO
In the code N is the number of points desired (surface area of sphere x surface point density to nearest integer), R is the radius of the sphere (atomic van der Waals radius + probe radius) and X is the output array of Cartesian point co-ordinates ( where (R, q, j) are the corresponding spherical polar co-ordinates).
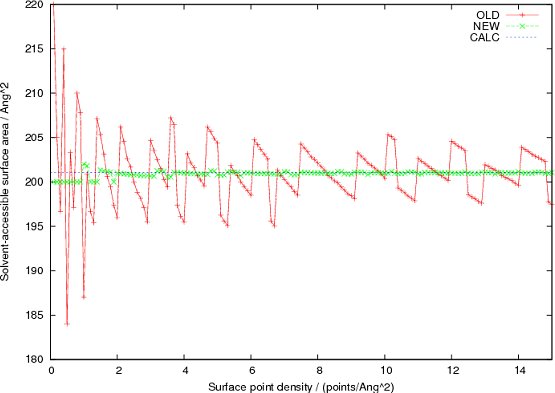
With this simple modification I get the results shown (labelled NEW), which are clearly much more accurate and more stable over a wide range of surface point density values. To be fair, the arrangement of atoms used in the test, with the interatomic vector along the z axis, is the worst possible case as then non-uniformity in the distribution at the polar regions where the atoms are in contact has the biggest effect. In real structures where the interatomic vectors are essentially in random directions, the average error is undoubtedly lower. With the new version a surface point density of 15 points/Ĺ2 seems to provide a reasonable trade-off between accuracy and computation time, so the default value has been reset to this.
The updated version of AREAIMOL will be in the next release of CCP4.
Briggs,P.J. (2000). CCP4 Newsletter No. 38, CCLRC, Daresbury, http://www.ccp4.ac.uk/newsletters/newsletter38/03_surfarea.html
Saff, E.B.& Kuijlaars, A.B.J. (1997). The Mathematical Intelligencer, 19, 5-11, http://www.math.vanderbilt.edu/~esaff/texts/161.pdf