| "What's New In CCP4 6.0" |
CCP4 Newsletter 44 Summer 2006 |
What's New In CCP4 6.0
Peter Briggs
CCP4, CSE Department, CCLRC Daresbury Laboratory, Warrington WA4 4AD
1 Introduction
CCP4 version 6.0 is the most recent major release of the CCP4 software
suite and was made in February 2006. A subsequent patch release 6.0.1
was made in June to fix a number bugs, however there were no major changes
to functionality.
The full list of changes to the suite since the previous major
version (5.0) can be found in the
CHANGES file
distributed with the core suite.
As the CHANGES file focuses mainly on technical changes, the aim of this
article is to give a quick tour of the new and updated features of the latest
release in a more user-friendly manner.
The article has the following outline:
2 New download and install mechanisms for version 6.0
The current release sees the rollout of a new integrated download and
install mechanism for the software, which is intended to vastly simplify
the otherwise complication task of selecting packages for download via a
simple interactive web interface. It also provides mechanisms for easy
installation of the packages once they have been transferred to the user's
system, using installers appropriate to the operating system in question
- see the article on the new system
elsewhere in this newsletter.
Using these mechanisms means that it is possible to have CCP4 and the
other packages described below working on a machine in just a few
minutes. Alternatively, old-fashioned style FTP download is also still
provided. All the downloads can be accessed via the
CCP4 Downloads Pages.
3 Updates to the software suite
3.1 The components of CCP4 release 6.0
For release 6.0 it was decided to break the distribution of the suite
up into the following "packages":
- Core CCP4
- Phaser and CCTBX
- Chooch
- CCP4mg
- Coot
The breakdown is for practical purposes to do with availability,
dependencies and rate of change of the various pieces of software
- inclusion or exclusion from the "core" was a pragmatic decision
and not an indication of the "quality" or "importance" of a particular
package. In this context the "core CCP4 package" essentially refers
to the the minimal CCP4 suite containing the CCP4 libraries plus the
standard programs, the user interface CCP4i, the Clipper software, and
some general "third-party" libraries.
Each of the new and updated packages are described briefly in the
sections below.
3.2 Updated software in the core CCP4
3.2.1 PDBCUR for coordinate file manipulations
PDBCUR
is a utility program for manipulating coordinate files, similar to
PDBSET but already with a large number of useful complementary functions
(for example selecting one model from a file which contains many models).
Additional functions new for CCP4 6.0 include:
- Produce a summary of the file PDB contents (for example, the number
of models, the number of chains and their composition)
(SUMMARISE keyword)
- Remove all hydrogen atoms from the model (DELHYDROGEN) *
- Select the most probable alternate conformation found in the file,
defined as that with the highest occupancy (MOSTPROB). *
- Remove all atoms with occupancy below a specific threshold
value (CUTOCC) *
- Move a subset of atoms in the model (TRANSLATE)
The options marked with * are also available via the CCP4i interface, in the
Edit PDB File task in the Coordinate Utilities module
(note that you must select the pdbcur option in order to access
the new functions).
3.2.2 MATTHEWS_COEF for data analysis
MATTHEWS_COEF
is traditionally used to estimate the number of molecules in the unit
cell based on solvent content. In CCP4 6.0 it has been updated to
additionally output the Kantardjieff and Rupp resolution-based
probabilities for each possible number of molecules (Protein
Science 12 1865-1871 (2003)), providing an additional
tool for estimating the contents of the cell.
The corresponding CCP4i task Cell Content Analysis in the
Molecular Replacement module has also been updated accordingly.
3.2.3 PDB_EXTRACT for structure deposition
The RCSB PDB_EXTRACT
program has been upgraded since CCP4 5.0, to version 1.7 in release 6.0 and
more recently version 2.0 in 6.0.1. PDB_EXTRACT can automatically extract data
from program log files and other output as the structure determination
progresses, and thus help to ease the burden of the final deposition
process (see
the
article in the previous newsletter on this and other RCSB PDB Tools).
PDB_EXTRACT is also accessible from within CCP4i via the Data Harvesting
Management Tool in the Validation and Deposition module.
For more information about PDB_EXTRACT and other RCSB PDB Tools, see
http://sw-tools.pdb.org/.
3.2.4 Other significant updates
The core CCP4 has a number of other updates to existing programs, most
significantly:
- REFMAC 5.2.0019
- MOLREP 9.0.09
- SFCHECK 7.0.18
- MOSFLM 6.2.6 (was 6.2.5 in CCP4 6.0)
3.3 New Programs in the core CCP4
There are a number of new programs in the core CCP4 package in 6.0:
- SUPERPOSE: secondary-structure-based coordinate alignment
- BP3: heavy atom phasing & refinement
- CHAINSAW: molecular replacement model preparation utility
- PIRATE and the Clipper utilities: statistical phase improvement
and related software
- PDB_MERGE: utility for combining the content of two PDB files
Each of these is described in more detail in the following sections.
3.3.1 SUPERPOSE
SUPERPOSE is
a secondary-structure based structural alignment program. It is an
alternative to the least-squares fitting method used in LSQKAB - SUPERPOSE
uses the secondary structure features to perform the initial fit, followed
by alignment of the protein backbone Cα atoms.
The program takes two coordinate files as input and outputs the transformation
matrix required to map one (the "moving" coordinate set) onto the other (the
"fixed" set). It also gives a per-residue listing of the quality of the match,
and identifies the secondary structure.
SUPERPOSE can be run from within CCP4i by selecting the ...Secondary
Structure Matching option in the Superpose molecules task (in the
Coordinate Utilities module). It is also used as the structure alignment
engine within both CCP4mg (under Applications -> Superpose
proteins) and Coot (under Calculate -> SSM
Superpose...).
3.3.2 BP3
BP3 is a heavy atom
refinement and phasing program which uses a multivariate likelihood approach that
has been reported to perform well in tests compared with other substructure
refinement programs (see the references in the
CRANK/BP3 article in an earlier newsletter).
It can be applied to various types of experiment including single- and multiple
isomorphous replacement (SIR and MIR), single- and multiple-wavelength anomalous
dispersion (SAD and MAD) and various combinations thereof (i.e. SIRAS and MIRAS
experiments).
Another advantage BP3 offers over CCP4's workhorse substructure
refinement and phasing program MLPHARE, is that where MLPHARE forces the
user to treat MAD phasing experiments with a "pseudo-MIR" approach
(effectively selecting one wavelength as the "native" and then treating
the other wavelengths as "derivatives"), BP3 treats each anomalous
dataset equally and so has a better treatment of the phase errors.
BP3 can be run from CCP4i via the dedicated Bp3 - Phasing task
in the Experimental Phasing module, or as part of the automated
CRANK task in the same module (CRANK is described further
on in this article).
More information on BP3 can also be found at the
BP3 webpage.
3.3.3 CHAINSAW
CHAINSAW is a
molecular replacement preparation utility that mutates a template PDB file
using a previously-generated sequence alignment provided by the user. CHAINSAW
preserves parts of the template which are conserved in the sequence alignment
(and which are therefore more likely to be conserved in the target structure),
and prunes back other parts which are not conserved (and which are therefore
less likely to reflect the target).
CHAINSAW offers a number of different pruning methods (back to
gamma atom, back to beta atom, or back to last "common" atom) and preserves
more atoms than for example in a polyalanine model. It can accept sequence
alignments in a variety of different formats (including ClustalW, PIR and Blast
among others), which has the advantage that the user can optimise the alignment
using their favourite methods before feeding it into CHAINSAW.
CHAINSAW can be run from CCP4i from the Create Search Model
task in the Molecular Replacement module.
3.3.4 PIRATE and Clipper
PIRATE is a
statistical phase improvement program - crudely, it is a replacement for the
DM program. PIRATE uses phases from a previously-solved known structure
(called the reference structure) as input to the phase improvement process,
and doesn't require any a priori knowledgement of the solvent content of the
target. PIRATE is run from the command line using the cpirate
executable, or else through the Run Pirate task in the
Density Improvement module of CCP4i.
In tests conducted by the program's author PIRATE performed better than DM
in all but one case. It should be noted however that the program's performance
depends critically on the quality of the Hendrickson-Lattmann (H-L) coefficients
output from the phasing program used to generate the initial phases (as these
give an indication of the error in the input phases). The current version has
been "tuned" to work best with H-L coefficients output from PHASER
but is known to also work well with the output from SOLVE. Using the output
from MLPHARE will generally work less well, and tests with BP3 have not been
carried out yet.
PIRATE can also make automatic use of non-crystallographic symmetry (NCS),
if it is given a file of heavy atom coordinates.
For typical structures without any unusual features the choice of reference
structure is not critical provided that it is relatively large - therefore the
CCP4 distribution includes the structure factors for 1AJR in .na4 format
(so-called "ascii-MTZ" - to convert to MTZ proper use the NA4TOMTZ program, or
run the Convert to MTZ... task in the Reflection Data Utilities
module). For structures with more unusual features (for example metalloproteins
or RNA-complexes) it would be worth trying different reference structures
which more accurately reflect the features of the target - in this case PIRATE
includes a option to evaluate multiple reference structures (check Use
evaluation mode in the PIRATE interface, or specify the
-evaluate option if running cpirate).
The program MAKEREFERENCE is used to generate the reference input for PIRATE
(use the cmakereference executable, or the Make Pirate Reference
task in the Density Improvement module of CCP4i). MAKEREFERENCE requires
both coordinates and structure factors for the reference structure, and outputs
new coordinates and structure factors for input into PIRATE. If the computer
has an internet connection then this data can be automatically downloaded from
the PDB archive at the EBI.
The PIRATE program is built on top of a set of crystallographic
software libraries called Clipper. In addition to PIRATE
and MAKEREFERENCE, CCP4 6.0 and later also contain a number of
other Clipper-based utilities that perform small tasks such as
generating maps and comparing phases. The utilities can be accessed
from CCP4i via tasks in the Clipper Utilities module, and the
CCP4i documentation gives a
list of the
available programs.
Information about PIRATE, Clipper and the related software can be found in
Kevin Cowtan's webpages.
3.3.5 PDB_MERGE
PDB_MERGE is a
useful jiffy for combining models from different sources, for example:
constructing a complex from its components, constructing a new model of
a protein using domains from several other models, or adding in separately
generated symmetry mates. PDB_MERGE has just two modes (MERGE/NOMERGE), which
control whether chains are merged at the same time as merging the data in the
files.
3.4 Updates to CCP4i
CCP4i is the CCP4 graphical user interface system. The current
version (released in CCP4 6.0.1) is 1.4.4.1, and the major
changes have already been reported in a previous
newsletter article. These developments are
summarised in the following sections.
3.4.1 New core tools in CCP4i
The new core tools are illustrated in figure 1 and include:
|

Figure 1: a map of the new core CCP4i tools and features
|
- Greying out task buttons for certain tasks where the underlying
programs are not found on the user's path
- A tool to allow searching and sorting of the jobs in the project
database
- A function to allow the user to choose custom colours for the jobs
displayed in the project database, to enhance quick comprehension of
the content
- A button to allow quick switching between different projects
directly from the main window
- Top-level help split into a menu with a number of subtopics, to
help find relevant documentation more quickly
|
3.4.2 New interfaces
There are two major new interfaces:
CRANK
CRANK
is a suite of programs for automated macromolecular
structure solution. Currently Crank supports SAD, SIR and SIRAS
experiments and makes use of various new and existing programs,
including BP3, SHELX and various CCP4 programs.
CRANK starts from scaled and merged data and allows the
automatic solution of macromolecular structures up to the point of
density modification. However it also has a "transculent box" design,
intended to help teach novice users about the various programs used
in crystallography.
The CRANK interface is part of CCP4i's Experimental
Phasing module. A more detailed article about CRANK appeared in
a previous newsletter; alternatively, visit the
CRANK webpage
for further information.
Shelx C/D/E
The SHELX_CDE
task interfaces to the SHELX programs,
specifically SHELX C, D and E. The task takes either SCALEPACK format
reflection files (note that the SCALA task can now output
SCALEPACK-style files that are suitable for input into SHELX and SOLVE)
or MTZ files containing intensities (preferred) or structure factor
amplitudes as input.
The task can be used to run the programs in a "pipeline" fashion from
data preparation through heavy atom site location to density
modification and hand determination, and generates useful plots from the
output of each program.
Like CRANK, the SHELX_CDE interface is also part of CCP4i's
Experimental Phasing module. Note that the SHELX programs
themselves must be obtained directly from the
SHELX website.
3.5 The other new packages in CCP4 6.0
3.5.1 PHASER and CCTBX
PHASER is a maximum likelihood-based phasing program. The
current version distributed by CCP4 (PHASER 1.3) has methods for
molecular replacement, however functions for experimental phasing are
also under development. One of PHASER's strengths is that its scoring
function gives a more accurate estimate of the quality of its molecular
replacement solutions; another is that it can search for solutions using
ensembles made up of many possible search models, with each model's
contribution to the ensemble weighted by estimates of its similarity
to the target. It also allows searching for multiple molecules in
multiple spacegroups.
PHASER's functionality can be accessed in a variety of
ways - it allows each step in the MR process to be run independently,
alternatively there is an "automatic" mode which will run through the
whole process without user intervention. PHASER can also be
run via a CCP4i task interface in the Molecular Replacement
module.
PHASER depends upon the CCTBX crystallographic software libraries,
and these must also be installed before building PHASER from source
(they are not required for binary installations). For more information
about CCTBX see the
CCTBX pages on SourceForge.
For more information about PHASER, see the
PHASER
webpages.
3.5.2 CCP4mg: presentation graphics in CCP4
CCP4mg is the official CCP4 molecular graphics package, and has
focused so far on structure analysis and high-quality presentation
graphics (including
movies).
CCP4mg is highly compatible with the CCP4 environment, and has an
interface with a similar look-and-feel to CCP4i.
CCP4mg is built around the idea of data objects
(the raw data that is loaded into the program, such as coordinates or
reflections) and display objects (the representations of
that data in the graphics window). A single data object can have
many associated display objects, which can represent all or just some
of the data (for example just the active site, a single chain or the
whole molecule) in a variety of different ways (for example as
ball-and-stick, ribbons, spheres or surfaces) and can reflect different
properties of the data (for example secondary structure elements,
solvent accessibility or electrostatic potential). Multiple display
objects are easily generated and combined to quickly build complex
representations.
Some examples taken from the CCP4mg website are shown in figures
2 thtough 4 below. These and other images showing the capabilities
of the program can be seen in the CCP4mg Gallery, and in the examples in the
10-minute
tutorials.
 |
 |
 |
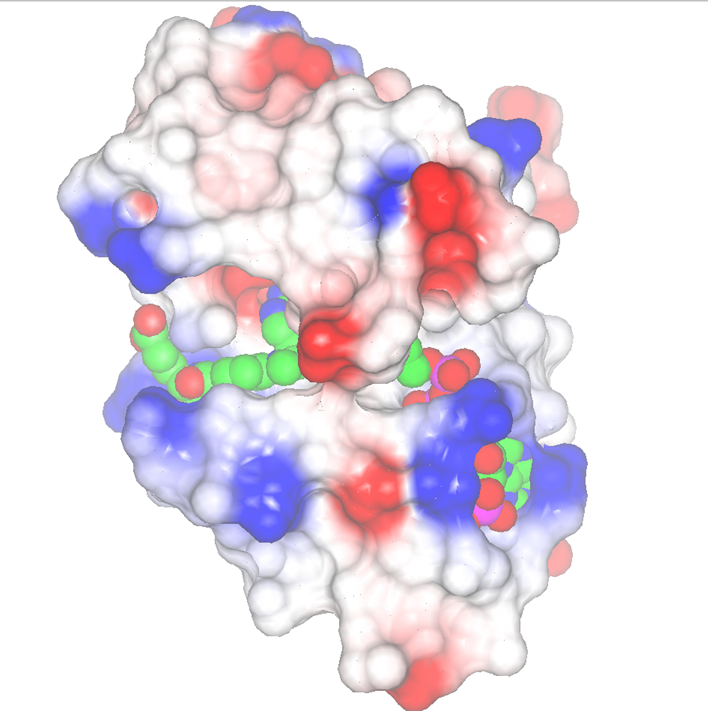
| Figure 2: Displaying a surface coloured by electrostatic potential |
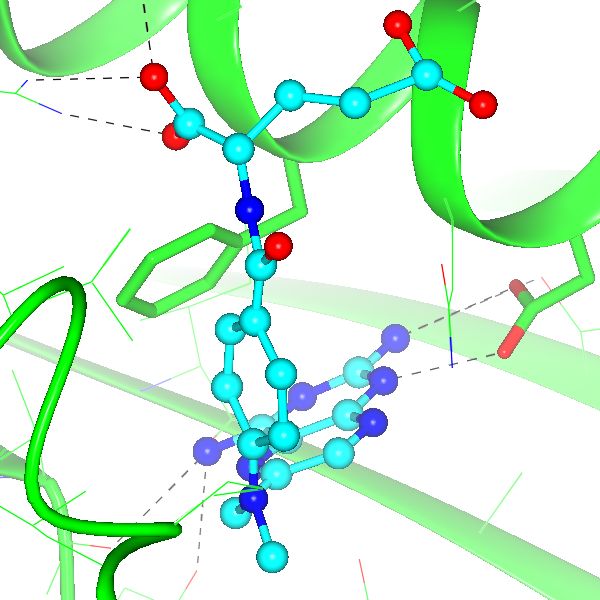
Figure 3: Active site in 1DFR, showing the local environment |
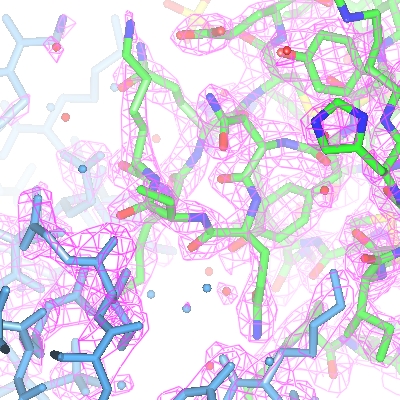
Figure 4: Displaying an electron density map |
| * Images taken from the
CCP4mg website. |
CCP4mg has support for generating molecular surfaces (including
transparent surfaces), and has various display modes for situations like
protein-RNA complexes and
ligand binding to DNA. The program can also
display maps
(for example to show
electron density), either directly from a CCP4 map file or generated "on the fly"
from an MTZ file containing structure factor amplitudes and phases. In addition
it has tools for
model superposition (via SUPERPOSE), generating symmetry and packing
diagrams, and for examining the local environment for part of a protein model.
There is also functionality for adding captions and legends, for customising
colours, and for rendering images at high-resolutions suitable for publication.
For more information see the CCP4mg
website.
3.5.3 Coot
Coot is a platform for semi-automated model building and
validation tools, and as such its functionality complements that in
CCP4mg. The validation tools in Coot (including
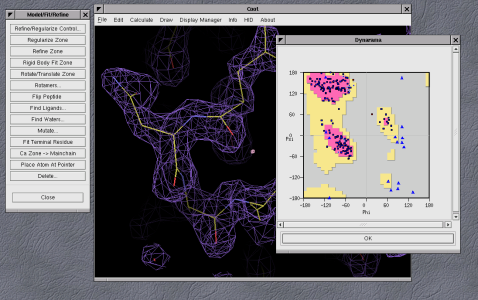
"dynamic" Ramachrandran plots as shown in figure 5, and geometry and
B-factor graphs) allow the user to quickly focus in on areas of the model
that are a poorly fitted, and once problems are identified there are a
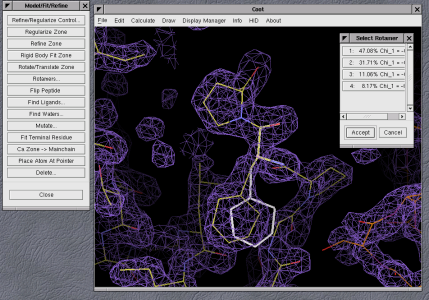
number of tools to help fix them (for example, rotamer fitting as shown
in figure 6, residue mutation, loop fitting and real-space refinement
amongst others).
Coot also offers tools to search for waters and for other
"blobs" of unmodelled density, and to then model them for example by
placing atoms in the density, or by allowing the user to pull ligands from
the REFMAC monomer library and "drop" them into the model. Coot also has
the option to run rounds of REFMAC refinement from within the program, as
well as a large number of other tools for model completion not described
here.
For further information about Coot visit the
Coot webpages.
 |
 |
| Figure 5: Example of Coot's dynamic Ramachandran plots |
Figure 6: Example of rotamer fitting in Coot |
| * Images taken from the
Coot website. |
3.5.4 CHOOCH
Having accurate values for the anomalous scattering factors f' and f''
is essential for the success of anomalous phasing methods. CHOOCH
is able to determine the values of these parameters from the raw
fluorescence spectra. Currently there is no CCP4i interface to
run CHOOCH, however this will be addressed in a future release.
An article on CHOOCH (in Word format) appeared in a previous
newsletter, or alternatively additional information is available
at the CHOOCH webpages.
4 Availability
Version 6.0 of the CCP4 software suite is available for download and use
free of charge for academic and non-profit use, upon the completion of a valid
licence agreement. The academic licence is included with the suite, or else
can be found via our licence
pages, however please note that the licence for version 5.0 is not valid
for version 6.0. Organisations wishing to use the software for commercial
purposes should contact CCP4 to obtain a commercial licence - this information
is also available via our licence pages.
Whenever possible we have endevoured to provide both source code and
precompiled binaries for all the programs and packages on all the platforms
that we have access to. This includes various flavours of Linux and some older
UNIXes, as well as Microsoft Windows and Mac OS-X. Our intention is to
give users of the software as many options as possible for installation.
The current major exceptions are CCP4mg and Coot, both of which are only
available from the CCP4 website as binary packages. This is for practical
reasons only: building these programs from source code is not a trivial task
and depends on the system setup and on the level of experience of the
installer. For those who do wish to take up the challenge, the source code
can be obtained from the appropriate websites:
http://www.ysbl.york.ac.uk/~ccp4mg/download/ for CCP4mg and http://www.ysbl.york.ac.uk/~emsley/coot/ for Coot.
5 Fixing bugs
As part of the life cycle of any software release, bugs are continually
being discovered and fixed. Fixes to problems are usually posted on the
Problems Pages, and
typically take the form of a "workaround" (suggested steps to avoid the bug)
or as fixes to the program source code. In this case the fixes are
normally distributed as "patches" (a patch is a file containing
instructions on how to modify another file, which can be read by the UNIX
patch program). It is always a good idea to check the Problems
Pages before reporting problems.
CCP4 6.0 and later includes a new utility called patch_ccp4.sh,
which on UNIX systems will automatically check for available patches to the
release, download and apply them. If your installation was from source
code then you still need to recompile any updated programs, however this
will also apply fixes to things like CCP4i which do not need to be
(re)compiled.
Depending of the lifespan of the release and the number of fixes
accumulated since the last release, patch releases such as 6.0.1 are
occasionally produced. These updates should not add any new
functionality. It is likely that at some stage there will be a 6.0.2
patch release of CCP4 6.0.
6 Promoting the current release
Since the release of CCP4 6.0, CCP4 staff and associated developers
have been visiting groups at various labs in the UK (and in one case further
afield) in order to demonstrate the new features of the software and talk
to scientists about using CCP4. Feedback from these visits has been
very positive, so if you feel that your group might benefit from a visit by
CCP4 staff and developers then please contact us at
ccp4@dl.ac.uk.
CCP4 staff are also attending a number of international conferences, with
exhibition stands at the ECM and ACA conferences amongst others, and will
be happy to demonstrate the software and to answer questions about it. Please
see the CCP4 courses pages
for the most up to date information.
7 Beyond 6.0: other programs and projects
Beyond CCP4 6.0 there are a number of other current and forthcoming
programs and projects associated with CCP4. Many of these are linked from
either the CCP4 Projects
Page or the Prerelease
Page.
Some of the programs that are now currently available outside of the CCP4
6.0 include:
MrBump: Molecular replacement with Bulk Model Preparation
MrBump is an automated molecular replacement pipeline, with emphasis
on generating a variety of search models. It uses various "helper"
applications (e.g. CHAINSAW), bioinformatics tools (e.g. FASTA) and
on-line databases (e.g. the PDB) to generate models, and then uses
MOLREP, PHASER and REFMAC for the MR steps.
In favourable cases MrBump has given a "one-button" MR solution; in
less favourable cases it can still be used to generate search models
for further investigation.
MrBump has a CCP4i task interface but can also be run standalone. It
can be obtained via
http://www.ccp4.ac.uk/MrBUMP/
and will be included in the next major release of CCP4.
PISA: Protein Interfaces Surfaces and Assemblies
PISA is an interactive tool for the exploration of macromolecular
(proteins and DNA/RNA) interfaces, prediction of probable quaternary
structures (assemblies) and database searches of structurally similar
interfaces and assemblies.
PISA is currently available as a webservice hosted by the MSD-EBI:
http://www.ebi.ac.uk/msd-srv/prot_int/pistart.html.
It is also planned that a future release of CCP4 will feature a
standalone version of PISA.
iMOSFLM
iMOSFLM is the new interface to the MOSFLM data processing program.
It is intended to replace the current dated X11-based interface with
a more intuitive and user-friendly graphical interface that will
also work on Microsoft Windows.
The GUI is now available for public download from http://alf1.mrc-lmb.cam.ac.uk/~geoff/mosflm/.
POINTLESS
POINTLESS does a number of things: it can be used to determine the
Laue group and spacegroup from a set of unmerged reflections; it
can score possible indexing schemes for a merged or unmerged dataset
against a merged dataset; and it can be used to reindex or change the
spacegroup of a reflection file.
POINTLESS can be obtained from the CCP4 Prerelease Page.
Buccaneer
BUCCANEER is a new Clipper application that performs statistical chain
tracing by identifying connected alpha-carbon positions using a
likelihood-based density target. It is essentially a model building
program, and follows on very naturally from the output of experimental
phasing steps and in particular from PIRATE. Tests by the program's
author suggest that it is complementary to ARP/wARP, since BUCCANEER
is relatively insensitive to resolution but very sensitive to the quality
of the phases and the estimates of their errors.
BUCCANEER can be obtained via http://www.ysbl.york.ac.uk/~cowtan/.
8 Acknowledgements
The CCP4 project is a collaborative effort and continues to thrive through
generous contributions of time, energy and software from members of the UK and
internation PX communities. Unfortunately time and space do not permit the
acknowledgement here of all these valuable contributions, however
a list of
acknowledgements is included in the current release, and acknowledgements
for the specific developments described in this article are given below:
- PHASER is developed by Randy Read's group (Wellcome Trust,
Cambridge). CCTBX is developed by the Computational Crystallography
Initiative (CCI) at Lawrence Berkeley, California.
- SUPERPOSE, PISA and PDBCUR are developed by
Eugene Krissinell (MSD-EBI). The new functionality added to PDBCUR
for CCP4 6.0 was written by Martyn Winn.
- MATTHEWS_COEF was originally written by Misha Isupov; the
Kantardjieff and Rupp analysis was added by Charles Ballard.
- PDB_EXTRACT is developed by Huanwang Yang (RCSB PDB).
- BP3 is developed by Navraj Pannu (Leiden University).
CRANK has been developed by Navraj and Steven Ness.
- CHAINSAW is developed by Norman Stein (Daresbury Laboratory).
- PIRATE, BUCCANEER and Clipper are developed by
Kevin Cowtan at (York University).
- PDB_MERGE is developed by Martyn Winn (Daresbury Laboratory).
- CCP4i is maintained and developed by the CCP4 group at Daresbury.
The Database Search/Sort, Project Switching and Job Colourisation
functions were developed by Francois Remacle. The SHELX_CDE interface
was written by Peter Briggs. The SHELX programs themselves are developed
by George Sheldrick and co-workers.
- CCP4mg is developed by Liz Potterton and Stuart McNicholas at
(York University).
- Coot is developed by Paul Emsley (York University).
- CHOOCH is developed by Gwyndaf Evans (DIAMOND Light Source).
- The new download and installation mechanism was developed by the CCP4
group at Daresbury, with Francois Remacle as the programming lead.
- MrBump is developed by Ronan Keegan and Martyn Winn, originally under the
auspices of the e-HTPX e-science project and now as part of CCP4.
- iMOSFLM is developed by Geoff Battye (MRC-LMB Cambridge).
- POINTLESS is developed by Phil Evans (MRC-LMB Cambridge).
The images for CCP4mg and Coot were taken without permission (and thus
with apologies) from the relevant websites. PJB would also like to apologise
to anyone who has been missed from these acknowledgements.
The CCP4 suite is maintained, developed and released by the CCP4 group
in the Computational Science and
Engineering Department at CCLRC Daresbury Laboratory, and comprises
Charles Ballard, Peter Briggs, Maeri Howard, Ronan Keegan, Francois Remacle,
Dan Rolfe, Norman Stein and Martyn Winn.
The CCP4 project is supported by the BBSRC, by income from commercial
distribution of the software, and by CCLRC Daresbury Laboratory. CCP4 would
also like to thank the many people past and present who support the project,
both with their time and with their contributions to the software suite itself
- without which the project would not be able to exist.
Peter Briggs, July 5th 2006