Bulk solvent
correction for yet unsolved structures
By A. Fokine & A. Urzhumtsev
Laboratory of
Crystallography and Modelling of Mineral and Biological Materials, UPRESA 7036
CNRS, University Henri Poincaré, Nancy I, 54506 Vandoeuvre-lès-Nancy, France
e-mail : fokine@lcm3b.uhp-nancy.fr
Abstract
The bulk solvent correction becomes
a routine tool for macromolecular structure determination because the low
resolution diffraction data contain an important structural information but
cannot be used without such correction. The most reliable of the existing
solvent correction methods is the flat solvent model. However, this method can
be applied only when an atomic model is already placed in the unit cell; this
is necessary in order to estimate two key parameters of the bulk solvent model,
ksol and Bsol . The
statistical analysis of values of these parameters for already resolved
structures shows that their fluctuation is relatively weak; as a rule the
outliers correspond to incorrectly determined parameters. Therefore, the mean
values of ksol = 0.35 e/Å3
and Bsol = 46 Å2 may be used when refined values
cannot be obtained thus extending essentially the limits of the application of
the flat bulk solvent model. In particular, such modelling allows to increase
drastically the signal in the translation search in molecular replacement.
1. Introduction
The macromolecular crystals contain
a large part of disordered (bulk) solvent whose contribution to low resolution
reflections is quite significant. An atomic model of a macromolecule without
the contribution of the bulk solvent cannot describe these low-resolution
diffraction data correctly. On another hand, these data are important to avoid
map distortion (Podjarny et al.,
1981; Urzhumtsev, 1991) and to refine efficiently and correctly the atomic
macromolecular model (Kostrewa, 1997). It can be thought also that these data
can greatly improve the resolution of the translation problem in Molecular
Replacement (MR) method because they are insensitive to reasonable errors in
the atomic positions and in the model orientation.
Among several methods allowing to
estimate structure factors of the bulk solvent (for a review see Jiang &
Brünger 1994; Badger 1997), the flat solvent model (Phillips, 1980; Jiang &
Brünger 1994) has been proven to be of a superior quality with respect to
others (Jiang & Brünger 1994; Kostrewa, 1997).
In this model the binary function M (solvent mask) is introduced which is
equal to 1 inside the solvent region and to 0 outside. The structure factors of
the bulk solvent are calculated as the scaled Fourier transform ℱ of this function :
Fsolv(ksol,Bsol)
= ksol exp(-Bsolsin2(q)/l2) ℱ(M) (1)
The unknown parameters ksol and Bsol
of the bulk solvent are chosen from the best fit of total calculated structure
factor Ftotal to experimental data :
G(ksol,Bsol) = S [ |Fobs| - |Fsolv(ksol,Bsol) + Fatoms | ] 2 à min (2)
where Fatoms
are the structure factors calculated from the ordered atoms. Therefore, the
knowledge of an atomic model of macromolecule already placed in the crystal is
necessary to estimate ksol
and Bsol , the key
parameters of the method.
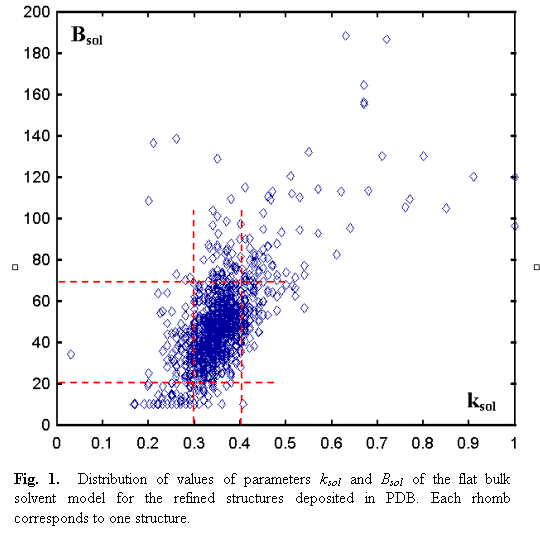
2. Statistical analysis of the bulk solvent parameters
In order to study the variability of
the values of ksol and Bsol we analysed their
distribution for the structures deposited in the Protein Data Bank (Bernstein et al., 1977). The corresponding models
have been selected using the provided software (3DB Browser; http://pdb-browsers.ebi.ac.uk/pdb-bin/pdbmain). The obtained distribution (Fig.
1) shows that for the most of structures the parameter ksol varies between 0.3 and 0.4 e/Å3 and Bsol varies between 20 and 70 Å2. The mean values
are equal to ![]() = 0.35 e/Å3 and
= 0.35 e/Å3 and ![]() = 46 Å2, and the dispersion are
0.03 e/Å3 and 17 Å2 , respectively (this statistic was
calculated for the models with 0< ksol <0.6 e/Å3
and 0< Bsol <100 Å2).
= 46 Å2, and the dispersion are
0.03 e/Å3 and 17 Å2 , respectively (this statistic was
calculated for the models with 0< ksol <0.6 e/Å3
and 0< Bsol <100 Å2).
A detailed study has been carried
out for some outliers with the experimental data available in PDB in order to
find the reason for such unusual values of the scale parameters. In all cases
the deposited parameters have been found to be incorrect, and the optimal values
obtained by us with the complete data set using the systematic search were in
the limits reported above.
For such small variation of the
scale parameters, the corresponding variation of the structure factors Fsolv(ksol,Bsol) is also relatively weak suggesting
that the mean values ![]() and
and ![]() can be used when the refined values of the
parameters can not be obtained. In particular, they can be used for the
molecular replacement when low resolution data are used as it is discussed
below. Another application is a map improvement when only a molecular envelope
is known (Fokine & Urzhumtsev, 2001).
can be used when the refined values of the
parameters can not be obtained. In particular, they can be used for the
molecular replacement when low resolution data are used as it is discussed
below. Another application is a map improvement when only a molecular envelope
is known (Fokine & Urzhumtsev, 2001).
It should be noted that the
distribution of ksol and Bsol is quite different from
that obtained for similar parameters of the exponential scaling model (Glykos
& Kokkinidis, 2000). This can be explained by a more poor quality of this
latter model, specially at a middle resolution (Urzhumtsev & Podjarny,
1995a), and by less clear physical meaning of the parameters of the exponential
model.
3. Bulk solvent correction and fast
translation search
It has been shown by Urzhumtsev &
Podjarny (1995b) that low resolution reflections being less sensitive
to model imperfection (including the errors in its orientation) can be
extremely useful for the solution of the translation problem. However, standard
molecular replacement protocols, except those by Glykos & Kokkinidis (2001)
where the exponential model is used, do not use reflections with the resolution
lower than 10-15 Å because they are strongly influenced by the bulk solvent.
For the translation search, the bulk solvent
correction eventually can be done at every position of the search model (while
the obtained ksol and
Bsol can be completely
unreasonable for wrong positions). Unfortunately, such way of solvent
correction cannot be included into fast translation algorithms (Navaza, 1994;
Navaza & Vernoslova, 1995) making its practical application inefficient.
However, the following observations can be done :
a)
For
the positions in the unit cell where the search model does not overlap with its
symmetrically related copies, the mask of the region occupied by all molecules
can be calculated as a junction of masks of individual molecules related by
symmetries; as a consequence, the structure factors of such total molecular
envelope can be rapidly recalculated from the structure factors of the envelope
of a single model;
b)
if the
structure factors of the envelope of a single model are preliminary scaled by ![]() and
and ![]() , such total
structure factors summarised over the symmetries give a good estimation of the
bulk solvent structure factors;
, such total
structure factors summarised over the symmetries give a good estimation of the
bulk solvent structure factors;
c)
for
all non-overlapping model positions, such scaled structure factors from the
envelope being added to the structure factors from the atomic model and
expanded over all symmetries are structure factors from the whole content of
the unit cell;
d)
as a
conclusion, a fast FFT-based translation search (Navaza, 1994; Navaza &
Vernoslova, 1995) done using bulk-solvent-corrected structure factors instead
of the values calculated
directly from the atomic model allows to compare correctly the magnitudes of
all reflections including those at low resolution for all non-overlapping
positions.
Naturally, spurious peaks in the translation function are eventually possible for the positions where the models overlap; however, these spurious peaks will be eliminated by the packing criterion and will not appear in the final list anyhow.
4. Tests protocols
A good approximation for the molecular
envelope can be available from a more or less complete atomic model. This is
the case when NMR models are used as the search models for the molecular
replacement. Several such cases reported as most difficult (for a corresponding
review see Chen et al., 2000; Chen,
2001) were chosen to test the suggested approach of the improvement of the
translation function (Table 1).
All test calculations were done with
experimental data, and the orientation of the search models was supposed to be
known (it can be noted that typical errors of about 5° in model orientation
practically did not influence the searches when low resolution reflections were
included). All translation searches were made with CNS (Brünger et al., 1998) using the fast translation
function (Navaza & Vernoslova, 1995). The translation search parameters
were taken without any optimisation (Chen at
al., 2000); complete NMR models were taken as they are in the PDB; the
B-factors for all atoms of the search models were assigned to be equal to 20 Å2.
In each test, a single NMR model was used for the translation search.
Table 1. Test structures : summary information
|
Protein name (reference) |
PDB ID / NMR ID |
Space group and unit cell parameters a,b,c (in Å) |
Percentage of the solvent in the unit cell |
|
Human interleukin-4 (Müller et al.,1995) |
1hik 1bcn |
P41212 92.1, 92.1, 46.4 |
63 |
|
P53 Tetramerization Domain (Mittl et al., 1998) |
1aie 1pet |
P422 45.5, 45.5, 32.2 |
53 |
|
Corn Hageman Factor Inhibitor (Behnke et al. 1998) |
1bea 1bip |
P42212 57.12, 57.12, 80.24 |
49 |
5. Results of improved translation
searches
Figure 2 shows the results of the
translation searches performed with and without low resolution data, with and
without the bulk solvent correction using three experimental data sets. Each
diagram shows the results of the translation search at a given resolution
shell. The top diagrams show the peaks obtained in the translation search
without any bulk solvent correction; the down diagrams show the peaks obtained
at the same conditions when the bulk solvent correction was taken into account
as suggested above. The height of each peak is shown in percents to the height
of the first peak of the corresponding search, and the correct solution is
indicated in red. It may be reminded that the total computation time for both
type of the translation function was the same due to the fast correction
procedure described in the previous section.
For human interleukin-4 (Müller et al., 1995), the translation search
performed at the standard resolution of 4-15 Å without solvent correction gave
the solution as the second peak. Including of all available reflections with
the resolution lower than 15 Å brought the correct peak to the first position.
Bulk solvent correction increased the contrast of the signal drastically,
specially when low resolution data were included (Fig. 2a).
For p53 tetramerization domain
(Mittl et al., 1998), the translation
search without solvent correction at standard 4-15 Å resolution gave the
correct solution hidden in noise and the search at 3-15 Å resolution gave it
slightly higher in the list. With the solvent correction, the peak for the
solution became the first with the best contrast at 4 Å even when no more low
resolution data are available (Fig. 2b).
CHFI (Behnke et al., 1998) was reported as the worst case among all NMR-based
searches (Chen et al., 2001). The
multiple rotation function (Urzhumtseva & Urzhumtsev, 2001) allowed to find
the orientation of the search model quite unambiguously and precisely and it
was supposed to be known for the translation search. Without the bulk solvent
correction, the solution did not appear among 10 highest peaks neither at the
resolution 5-15 Å nor at 4-15 Å. When all available magnitudes with the
resolution lower than 5 Å were used, the correct peak was the 7th in
height. At the same time, with the bulk solvent correction, this peak became
the first one for the resolution lower than 5 Å while the contrast is not so
high as for two previous cases (Fig. 2c).
Therefore, it can be concluded that
the bulk solvent correction using the flat solvent model improves drastically
the translation function, quite differently from the correction by the
exponential model where no significant improvement has been observed (Glykos
& Kokkinidis, 2001).
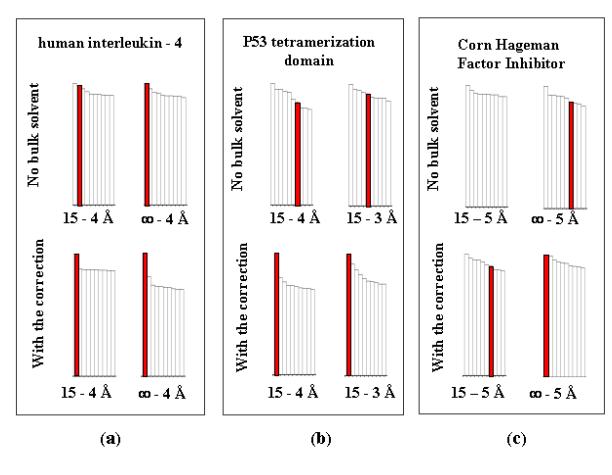
Fig. 2
Relative heights of peaks of the translation function
Each column represents a peak of the
translation function and has a height in percents to the value of the first
(highest) peak obtained at the same translation search. For each resolution
range ten highest peaks corresponding to searches without solvent
correction are shown at the top
diagram and ten highest peaks corresponding to searches with such
correction are shown at the bottom diagram. Peaks corresponding to the
correct solution are shown in red. The figures represent the results of the
searches for :
a)
human
interleukin-4 (Müller et al.,
1995),
b) p53
tetramerization domain (Mittl et al.,
1998),
c)
Corn
Hageman Factor Inhibitor (Behnke et
al., 1998).
|
Fig. 2
Relative heights of peaks of the translation function Each column represents a peak of the
translation function and has a height in percents to the value of the first
(highest) peak obtained at the same translation search. For each resolution
range ten highest peaks corresponding to searches without solvent
correction are shown at the top
diagram and ten highest peaks corresponding to searches with such
correction are shown at the bottom diagram. Peaks corresponding to the
correct solution are shown in red. The figures represent the results of the
searches for : a)
human
interleukin-4 (Müller et al.,
1995), b) p53 tetramerization domain (Mittl et al., 1998), c)
Corn
Hageman Factor Inhibitor (Behnke et
al., 1998). |
6. Physical meaning and possible values of the parameters of the flat solvent model
It has been discussed many times,
that the parameter ksol
describes the mean electron density of crystallisation solution. Kostrewa
indicated (1997) that the electron density of pure water is 0.33 e/Å3,
the density of 4M ammonium sulphate is
0.41 e/Å3, so normally the value of ksol should vary between these limits which corresponds
well to the distribution found from the PDB analysis (Section 2).
It is clear that the parameter Bsol describes the sharpness
of the solvent density at its border but his physical meaning has not been
discussed previously. The larger is Bsol
the deeper the electron density of the solvent penetrates to the macromolecular
region and therefore very large values of Bsol
are meaningless. On the other hand, the distance interval on which the electron
density of the solvent decreases to zero should be at least larger than the
radius of the solvent molecule (1.4 Å).
In fact, we have found that the
optimal value of this parameter corresponds to the mostly flat electron density
distribution at the border between the solvent and molecular regions (details
will be published elsewhere; manuscript in preparation). Again, these values
agree well with the distribution found statistically.
It can be noted that a non optimal
choice of parameters does not allow to fit equally well all calculated data to
the experimental values and usually leaves elevated R-factor for lowest
resolution reflections. In most of cases, such wrong choice can be avoided
either by a systematic search or by a local search for ksol and Bsol
as it is realised in CNS (Brünger et al.,
1998) but starting from ![]() and
and ![]() ,
differently from the currently existing procedure.
,
differently from the currently existing procedure.
7.
Conclusions
The distribution of values of the
bulk solvent parameters ksol
and Bsol for
crystallographic structures deposited in Protein Data Bank shows that their
correct values vary in relatively small limits around ![]() = 0.35 e/Å3 and
= 0.35 e/Å3 and ![]() = 46 Å2.
These limits and corresponding mean values have a reasonable physical
interpretation; ksol
corresponds to the means electron density of the solvent and the optimal value
of Bsol provides with the
smooth and flat transition of the electron density between the solvent and
molecular regions.
= 46 Å2.
These limits and corresponding mean values have a reasonable physical
interpretation; ksol
corresponds to the means electron density of the solvent and the optimal value
of Bsol provides with the
smooth and flat transition of the electron density between the solvent and
molecular regions.
For a known atomic model in the unit
cell, the optimal values of the bulk solvent parameters can be found either by
systematic or by a local search; in the latter case, the start from ![]() and
and ![]() allows to avoid a wrong answer.
allows to avoid a wrong answer.
When the standard procedure can not
be applied to obtain the optimal values of the parameters, for example when an
atomic model in the unit cell is not known yet, the mean values ![]() and
and ![]() can be used instead of the optimal values.
can be used instead of the optimal values.
In particular, this latter allows to
include the bulk solvent correction using the flat solvent model into fast
calculation of the translation function. The use of low resolution reflections
with such bulk solvent correction improves drastically the signal in the
translation search.
References
Badger,
J. (1997). Methods Enzymology. 277,
344-352.
Behnke,
C.A., Yee, V.C., Le Trong, I., Pedersen, L.C., Stenkamp, R.E., Kim, S.-S.,
Reeck, G.R. & Teller, D.C. (1998). Biochemistry, 37, 15277-15288.
Bernstein, F. C.,
Koetzle, T. F., Williams, G. J., Meyer, E. E. Jr., Brice, M. D., Rodgers, J.
R.,
Kennard, O., Shimanouchi, T. & Tasumi,
M. (1977). J. Mol. Biol. 112, 535-542.
Brünger, A.T.,
Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R.
W.,
Jiang, J.-S., Kuszewski, J., Nilges, M.,
Pannu, N. S., Read, R. J., Rice, L. M., Simonson, T.
& Warren, G. L. (1998). Acta Cryst., D54, 905-921.
Chen,
Y.W., Dodson, E.J. & Kleywegt, G.J. (2000). Structure, 8, 213-220.
Chen,
Y.W., (2001). Acta Cryst. D57,
1457-1461.
Fokine, A. & Urzhumtsev,
A. (2001). CCP4 Newslett. 39,
71-78.
Glykos, N. M. &
Kokkinidis, M. (2000). Acta Cryst. D56, 1070-1072.
Glykos, N. M. &
Kokkinidis, M. (2001). Acta Cryst. D57, 1462-1473
Jiang, J.-S. & Brünger, A.
T. (1994). J. Mol. Biol. 243,
100-115.
Kostrewa, D. (1997). CCP4
Newslett. 34, 9-22.
Mittl, P., Chène, P. & Grütter, M.G. (1998). Acta Cryst. D54, 86-89.
Müller, T., Oehlenschläger, F. & Buehner, M. (1995) J.Mol.Biol., 247, 360-372.
Navaza,
J. (1994). Acta Cryst. A50, 157-163.
Navaza, J. & Vernoslova, E.
(1995). Acta
Cryst., A51, 445-449.
Phillips, S. E. V.
(1980). J.Mol.Biol., 142, 531-554.
Podjarny, A. D., Schevitz, R.
W. & Sigler, P. B. (1981). Acta Cryst. A37, 662-668.
Urzhumtsev, A. G. (1991). Acta Cryst. A47, 794-801.
Urzhumtsev, A.G. & Podjarny,
A.D. (1995a) CCP4 Newslett. 31, 12-16.
Urzhumtsev, A.G., Podjarny, A.D.
(1995b) Acta Cryst., D51, 888-895
Urzhumtseva, L. & Urzhumtsev,
A.G. (2001) CCP4 Newslett. 39,79-85