
Stephen J. Maginn, on behalf of the staff of the Cambridge Crystallographic Data Centre (CCDC)
CCDC, 12 Union Road, Cambridge CB2 1EZ, UK
maginn@ccdc.cam.ac.uk
The Cambridge Structural Database (CSD) System is a well-known and widely used resource in structural chemistry. The Cambridge Crystallographic Data Centre (CCDC), which collates and makes the CSD System available, has recently been exploring the value and application of knowledge implicit within the database knowledge about molecular conformation and about intermolecular interactions.
Currently (forthcoming October 2001 release) containing 245392 entries, the CSD is the worlds repository for small molecule organic and metal-organic crystal structures. There is a strict definition of the turf covered by the CSD with respect to the Protein DataBank (PDB) structures with less than 1000 atoms in the asymmetric unit go into the CSD. CCDC has deposition arrangements with a host of journal publishers, whereby structures going through the publication process have their crystallographic data deposited at Cambridge, and once publication occurs, they are added to the database. An increasing number of unpublished structures, labelled as Private Communications, are now also included, and submission of these is encouraged, although all go through the same rigorous checking procedures as structures intended for publication.
The information stored in the CSD for each entry can be considered in three classes. Firstly, there is the text-based (and sometimes numeric) information, containing the bibliography (i.e. full literature reference, where appropriate), chemical names and formulae, some experimental information about the crystal structure determination procedure, and any other information that may be available (e.g. compounds use, colour and shape of crystals, etc. etc.). Secondly, there is chemical connectivity information in the form of a 2D structural diagram it is this that forms the basis of much of the sophisticated search mechanisms for the CSD System (see later). Thirdly, there is the crystallographic information, consisting of unit cell dimensions and space group, and atomic coordinates (these are available for the vast majority of entries, although not all). It is in this third category where the true value of the Database lies.
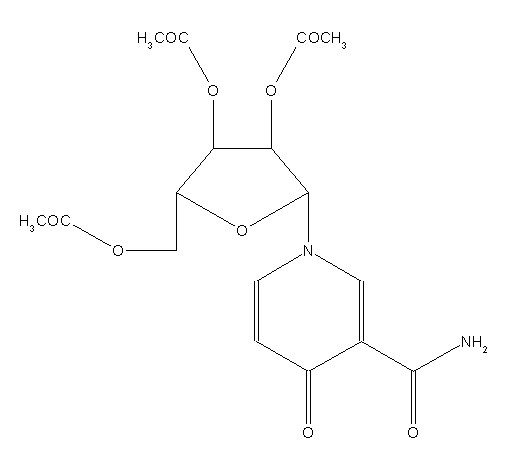
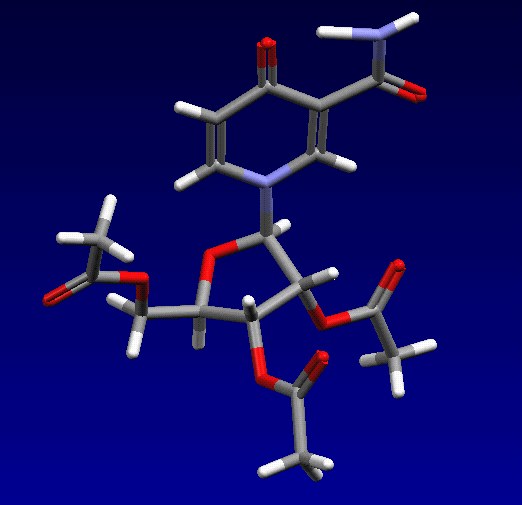
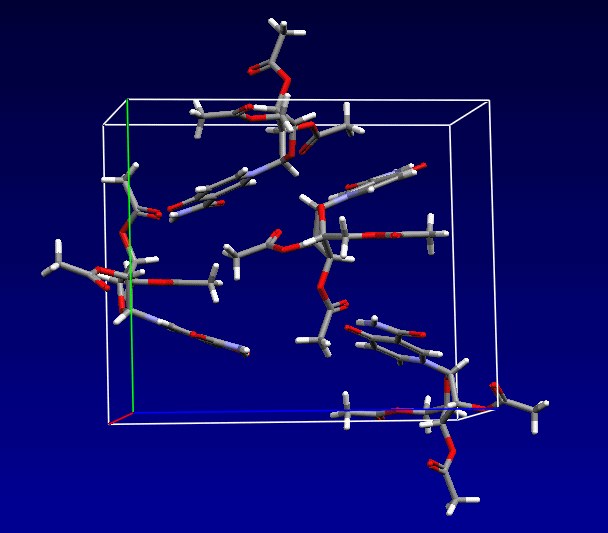
All database entries are identified uniquely by a six-letter reference code, or refcode, which may be followed by two digits if the entry is a member of a family of entries. Fig. 1 shows some of the information stored for the entry BASYOJ, for example.
Fig.1: Cambridge Structural Database contents: bibliographic, 2D structure, 3D coordinates and packing
The CSD is not provided to users in isolation. It comes with a package of software, designed for search of the Database and analysis of results, which can be used to extract knowledge about molecular conformation or intermolecular interactions.
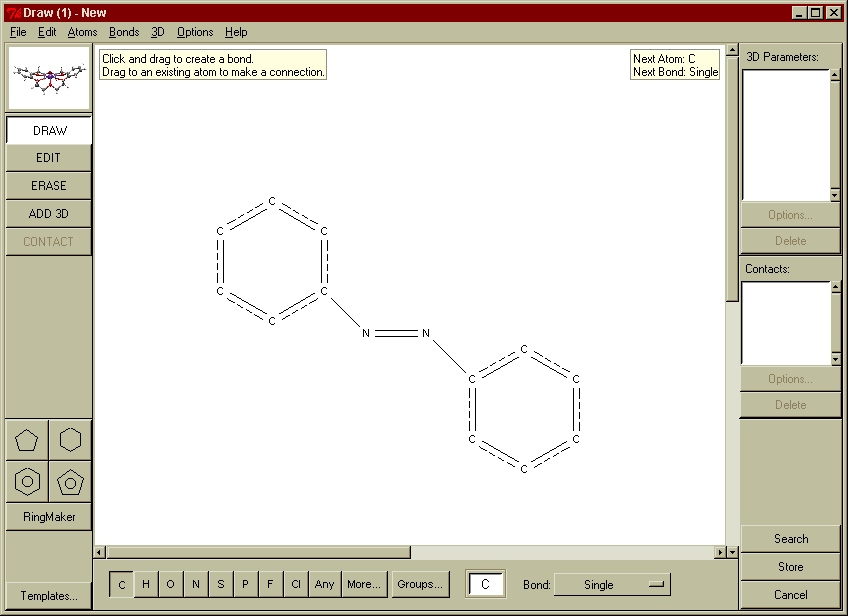
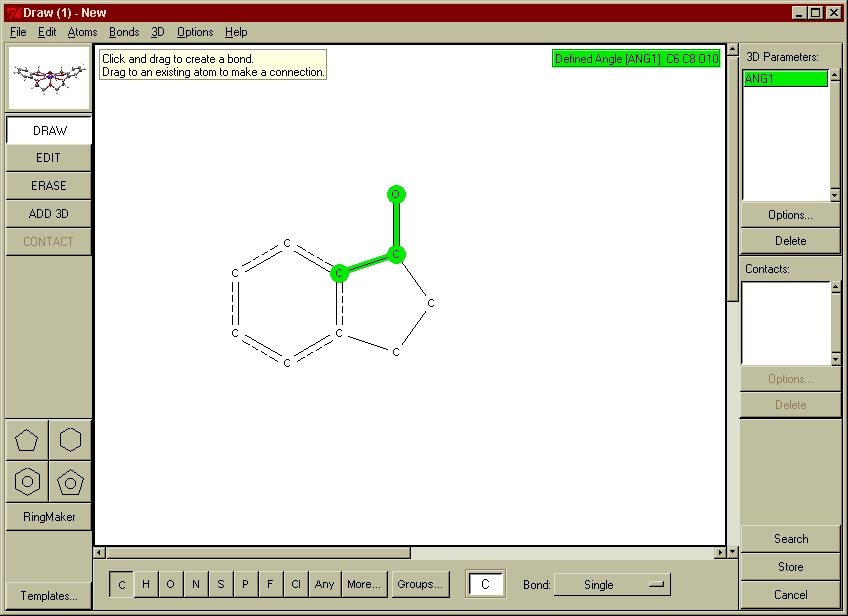
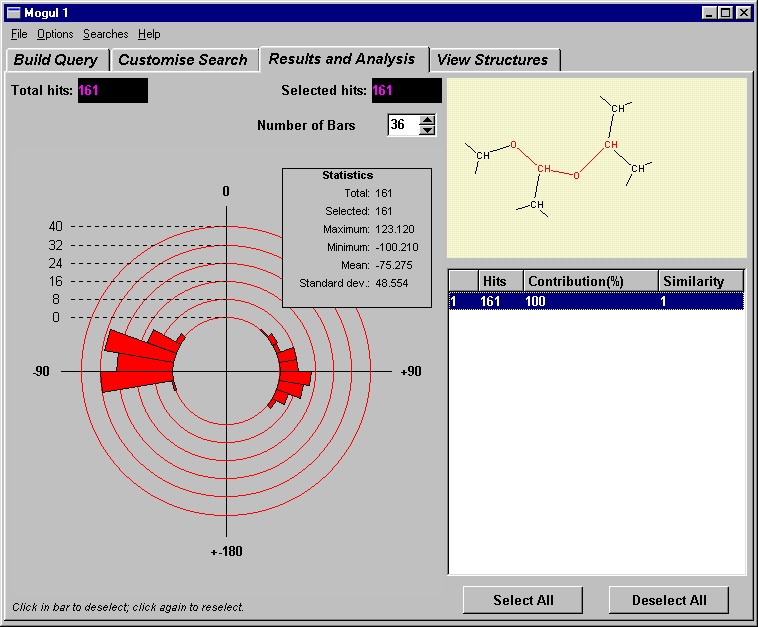
For many years, the main search interface to the CSD was the QUEST software. This remains available (see below), although the program has now been replaced by a much more modern interface known as ConQuest (see below for availability). All fields of data within the CSD are searchable using the ConQuest software, although the search functions used to extract geometrical knowledge are those based on the 2D structural diagram. Database search queries may be constructed to search for user-defined molecular fragments, within which molecular and intermolecular geometries may be tabulated and used to further constrain the search if required (see Fig. 2).
Fig. 2: A couple of examples of 2D fragment searches in ConQuest
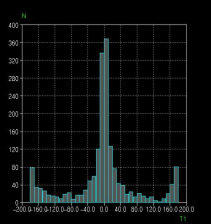
Once geometries have been tabulated, the information may be exported to Excel for analysis, or to the program VISTA, which also forms part of the CSD System. Trends in molecular geometry or intermolecular interactions for compounds or molecular fragments of interest may then be discerned and correlated (Fig. 3).
Fig. 3: Correlation of O=C-C-O torsion angles with the central C-C bond length. When there is a hydroxyl group involved, an intramolecular H-bond is set up (as is tautomerism) and the C-C bond shortens. Plots produced using VISTA.
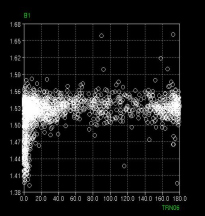
CCDC has decided to expand the capabilities of the CSD System by including two knowledge-based libraries. These encapsulate much of the information on intermolecular interactions and on molecular geometry found within the CSD, which has been pre-extracted and is presented to the user in an easy to visualise form. One of these libraries, IsoStar, has been available for some years, whereas the other, Mogul, is in development with a view to release in 2002.
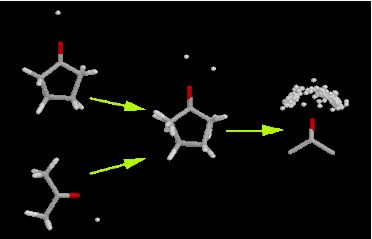
IsoStar [1] is a library of the intermolecular interactions found within the CSD and some from the PDB (see later). Searches of the CSD have been carried out for over 12000 particular intermolecular interactions. One of the participating fragments in each interaction has been designated a central group, the other an interacting group. The central groups are then superimposed and normalised for all the hits in each search, to produce scatterplots in which the interacting groups are arrayed around the central group, thereby displaying preferred geometries of interaction. It is possible in IsoStar to link back from individual data points in a scatterplot to the CSD entry that gave rise to that point.
Fig. 4: Methodology of IsoStar scatterplot production
IsoStar scatterplots can sometimes appear confusing, when there are thousands of hits contributing to them. The scatterplot can therefore be displayed as a contour plot, where the contours are values of density of interactions per unit volume of space. These may be scaled differently, by dividing by the expected density of interactions should they have been randomly distributed, such that they become probability or propensity densities, and can be directly compared (and combined see later) quantitatively as well as qualitatively (see Fig. 5).
Fig. 5: The scatterplot for hydroxyl interactions with an ester link, and its corresponding contour plot, revealing the preference for interaction with the carbonyl oxygen and its lone pair directionality.
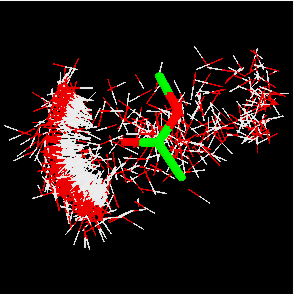
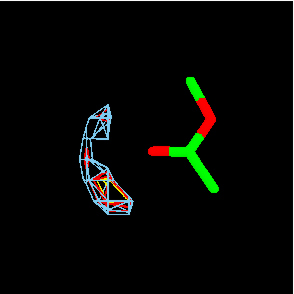
IsoStar also contains information on intermolecular interactions extracted from the PDB, in the form of over 3000 scatterplots with certain restrictions. The PDB interactions within IsoStar are between a ligand and its host protein, and only from structures determined at better than 2.5Å resolution. There are therefore nowhere near as much data contained within the PDB-derived plots as in the CSD-derived plots, and hydrogen atom positions are missing, leading to some loss of directional information. Nevertheless, this enables some comparisons between plots derived from CSD and PDB to be made, and some conclusions to be drawn about how appropriate it may be to use small molecule crystal data to model protein-ligand interactions (Fig. 6).
Fig. 6: Scatterplots for O-H and N-H interactions with a COO- central group, derived from the CSD (left) and PDB (right). Lone pair directionality is visible in both.

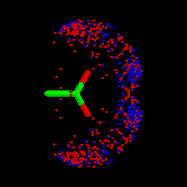
IsoStar also contains the results of some calculations of interaction energies for certain systems, produced using intermolecular perturbation theory [2], and model molecules.
IsoStar scatterplots and contour plots are displayed in a customised version of Rasmol, whereas the IsoStar framework itself is browser-based.
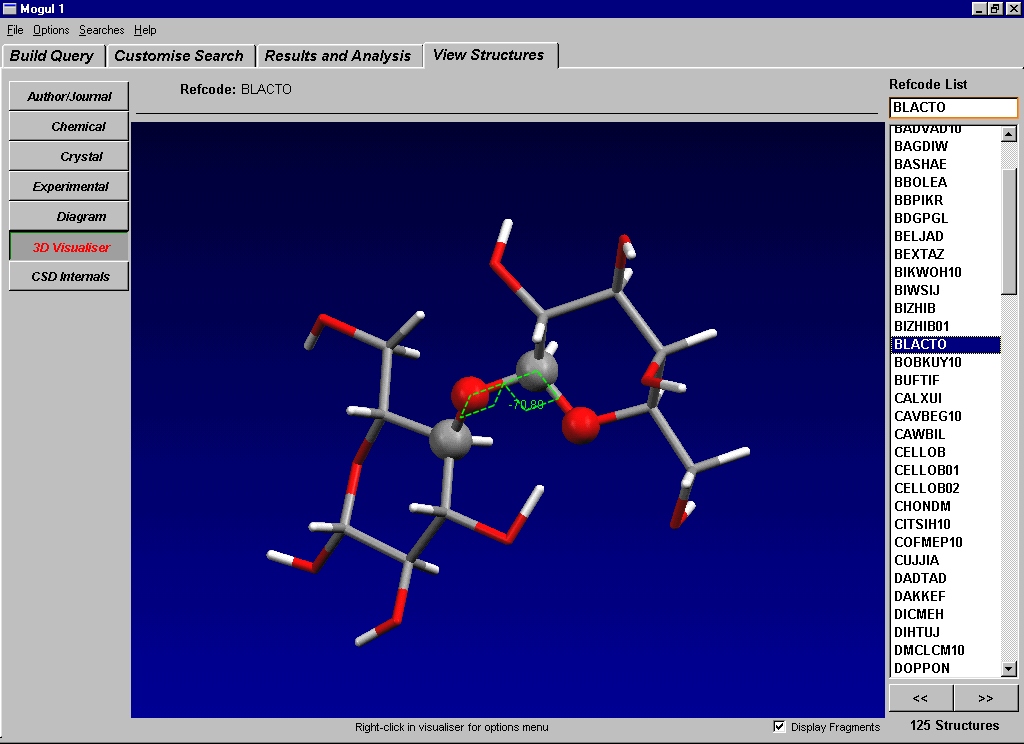
Currently in development at CCDC, Mogul embodies the molecular geometrical information from the CSD into three constituent libraries one of bond lengths, one of valence angles, and one of torsion angles (excepting ring torsions). Values of these parameters within molecules are heavily influenced by the chemical environments of the constituent atoms, and therefore atoms of the same element and hybridisation need to be more clearly defined than in traditional atom-typing arrangements. Atoms in Mogul are therefore exactly defined by going out to 2 bonded atoms away from each constituent atom, considering atom and bond types for each. When searches for an exact match with the studied parameter, to this level of definition, are carried out, one often finds that there is not enough data (i.e. not enough exact matches, or hits), even throughout the quarter of a million CSD entries, to be statistically significant enough to draw firm conclusions. However, the Mogul libraries have a hierarchical tree structure, so that the strict atom definitions can be relaxed, and one can move up the branches of the tree structure, in order to obtain enough information.
Mogul is expected to be ready for release as part of the CSD System in 2002.
Fig. 7: Use of Mogul to find the torsional distribution about a link between rings in a sugar molecule.
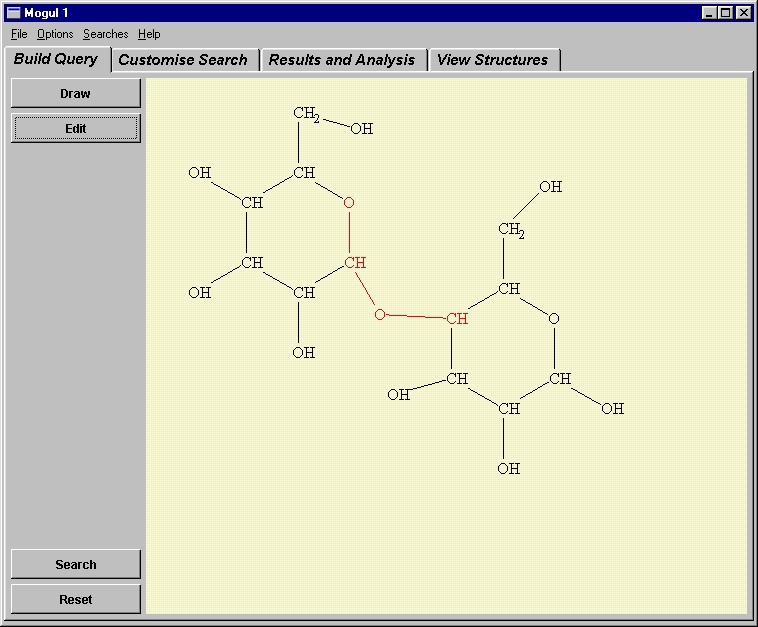
IsoStar and Mogul therefore represent a catalogue of knowledge, extracted
from the CSD (and in IsoStar's case also the PDB); such knowledge may also
be extracted by judicious use of the CSD and its accompanying software.
A similar procedure may be followed for extraction of conformational and
interactional knowledge from the PDB, for protein-ligand complexes, using
Relibase[3]. All this information is not merely of interest - it
is genuinely useful in approaching real-world problems.
It is possible to envisage many applications of both forms of crystallographic knowledge referred to above. There are obvious examples such as the use of conformational information in conformational search programs, or to compare with and/or validate the results of molecular modelling calculations. Crystallographic information may be able to feed back into the latter in the form of enhancements and alterations to molecular mechanics forcefields. There are other, less obvious applications though, in "bootstrapping" crystal structure determinations - for example, it may be possible to use the information in a more automated way to help protein threading in poor resolution electron density maps (similar to the use of Ramachandran plots), or to determine potential ligand conformations in binding sites where the density may be inconclusive. In small molecule structures, validation of new structures and structure solution from powder diffraction (where crystallographic information is added to a model to constrain the number of variable parameters and therefore give a better chance of structure solution from the limited data available) are but two potential applications. CCDC is involved in collaborations to explore some of these areas, but the applications which are perhaps best developed up to now, because of commercial interests, are those involving protein-ligand docking studies. Two examples are shown below, each using a different methodology of exploiting information extracted from the CSD (and PDB).
The program SuperStar [4,5,6] has been developed as a wholly knowledge-based approach to determining where particular organic functional groups (known as "probe groups") like to sit in macromolecular binding sites. A study of a particular binding site with a range of probe groups may therefore produce the necessary information required for the creation of a pharmacophore or as the first stage in an ab initio ligand design procedure.
SuperStar depends entirely upon the propensity plots (i.e. the scaled contour plots) contained within IsoStar. The program derives the identity of organic functional groups on the protein structure which intrude upon the surface of the binding site, retrieves the relevant plots from IsoStar where the protein's group is the central group and the user-selected probe group is the interacting group, and combines them (by multiplication, as we are considering propensities / probabilities) to produce a grand, 3D binding map within the protein binding site for the probe group.
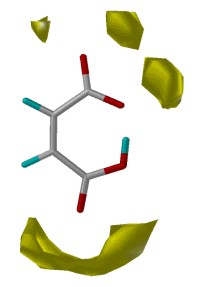
Fig. 8: Combination of IsoStar plots in SuperStar
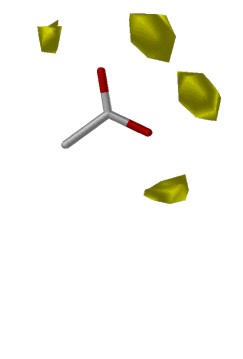 ×
× 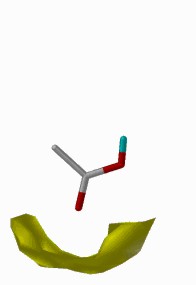 =
= 
The earliest versions of SuperStar have used only the IsoStar plots extracted from the CSD as their source. A new version is about to be released, however, which offers the choice of using the PDB-derived IsoStar plots instead; they are somewhat less well-defined as they contain less data, but it may be considered more appropriate to use them than CSD-derived plots in certain cases. SuperStar also has a limited capability for handling active site flexibility in that rotation of hydroxyl groups are considered.
This wholly knowledge-based approach employed by SuperStar offers huge advantages in terms of speed over a more traditional, energy-based approach to deriving this information in that there are no long-winded calculations. The academic papers [4,5,6] contain much in the way of validation of the method.
GOLD stands for Genetic Optimisation for Ligand Docking [7,8]. Unlike SuperStar, GOLD is not a purely knowledge-based program - it depends upon a forcefield to perform the core of its function, which is the derivation of preferred binding modes for particular ligands within particular binding sites. A genetic algorithm is the means used to optimise this - hence the program's name. However, the forcefield is enhanced by the use of crystallographic information in two key ways; firstly, ligand torsion angles can be restricted to CSD-observed ranges of values using a cruder version of the torsion library in Mogul; secondly, there is directional information from IsoStar, particularly concerning hydrogen bonding, hard-coded into the forcefield. Thus GOLD may be considered to be something of a hybrid between energy-based and knowledge-based methods. The methodology has been greeted with some acclaim in comparative studies [9].
Originally created as a 3-way collaboration between the University of
Sheffield (Dept. of Information Studies), former GlaxoWellcome and CCDC,
GOLD has been available for some time now and is constantly developing
- indeed, CCDC has recently entered into collaboration with Astex Technology
to facilitate onward development. GOLD's uses are in understanding how
molecules of known activity may bind to their target proteins, in the absence
of crystallographic information, and in so-called "virtual screening" studies,
where comparisons with a training set of experimental results may be used
to draw inferences about likely activity of compounds within a combinatorial
library.
Although it is early days in the exploitation of
crystallographic knowledge, derived both from the CSD and PDB, it is clear
that the potential applications are many and varied and the potential benefits
huge. Evidence seems to show that rather than replacing energy-based means
of modelling systems, these methodologies will enhance them in a symbiotic
way.
The Cambridge Structural Database, accessible via search programs ConQuest or QUEST, together with programs VISTA and PLUTO and the knowledge-based library IsoStar, is available free of charge to UK academics on the EPSRC-funded, Daresbury-based Chemical Database Service (CDS). For further details, see the CDS website.
Alternatively, the CSD System can be obtained for an annual subscription
fee direct from CCDC. GOLD and SuperStar are also available commercially.
See the CCDC website for details.
[1] I.J. Bruno, J.C. Cole, J.P.M. Lommerse, R.S. Rowland, R. Taylor, M.L. Verdonk; Journal of Computer Aided Molecular Design 11 (1997), p525-537
[2] I.C. Hayes, A.J. Stone; J. Mol. Phys. 53 (1983), p83-105
[3] http://relibase.ccdc.cam.ac.uk , http://relibase.ebi.ac.uk , http://relibase.rutgers.edu
[4] M.L. Verdonk, J.C. Cole, R. Taylor; J. Mol. Biol. 289 (1999), p1093-1108
[5] M.L. Verdonk, J.C. Cole, P. Watson, V. Gillet, P. Willett; J. Mol. Biol. 307 (2001), p841-859
[6] D.R. Boer, J. Kroon, J.C. Cole, B. Smith, M.L. Verdonk; J. Mol. Biol. (submitted)
[7] G. Jones, P. Willett, R.C. Glen; J. Mol. Biol. 245 (1995), p43-53
[8] G. Jones, P. Willett, R.C. Glen, A.R. Leach, R. Taylor; J. Mol. Biol. 267 (1997), p727-748
[9] C. Bissantz, G. Folkers, D. Rognan; J. Med. Chem. 43 (2000), p4759-4767