Department of Chemistry,
University of York,
Heslington,
York, YO10 5DD, U.K.
yao@ysbl.york.ac.uk
A number of protein structures have been solved and/or refined at atomic resolution since strong X-ray sources from synchrotron radiation and modern data collection techniques are available. With atomic resolution data ACORN [1] can solve a protein structure from a small fragment as little as 5% or even 1% of of the scattering matter of the unit cell.
The starting fragment can be found from various sources according to the features of the structure to be determined such as single random atom, heavy atoms (Sulphur or heaver), Alpha Helices or motif from other structures in Protein Data Bank (PDB). Since the size of a fragment is very small it is easy to find out a motif from PDB which is growing larger quickly.
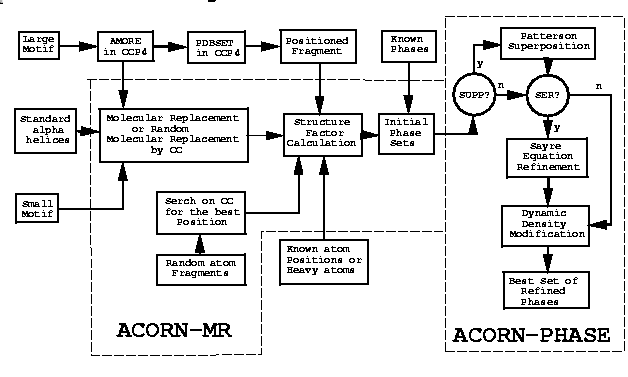
ACORN is divided into two parts: ACORN-MR and ACORN-PHASE. ACORN-MR is a fragment handling and generating a number of sets of initial phases with weights. ACORN-PHASE is a phase development and refinement procedure to obtain the best set of phases indicated by Correlation Coefficient (CC). Normally a model can be build up automatically from ACORN map by ARP/wARP in CCP4 or by QUANTA.
All reflections are divided into three groups: strong, medium and weak. The strong and weak reflections are used in phase refinement procedures while the medium reflections are used only for calculation of CC as a figure of merit to indicate solutions.
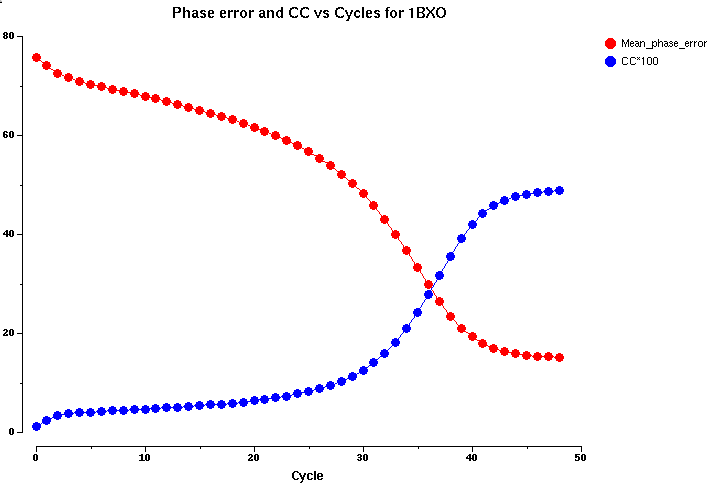
Correlation coefficient is computed between the normalized structure factors and calculated ones from fragment or from modified map. CC for medium reflections (CC-medium) is used to indicate the quality of the phases computed from a modified density map while this map is calculated using the strong reflections only. Here is an example to show the relation between CC-medium and phase errors vs the cycles of DDM and the CC-medium does indicate the solution clearly.
CC-medium and phases error:
CC for all reflections (CC-all) is used by a molecular replacement method in ACORN-MR to indicate correct orientation and position. CC-all is also used to indicate correct position for single random atom searching.
The first procedure in ACORN-MR is single random atom searching in a suitable region in unit cell according to the space group. ACORN-MR will generate a number of sets of single random atom and calculate CC-all for each set. Then all sets will be sorted on CC-all and provide maximum 1000 sets of initial phases with highest CC-all to ACORN-PHASE. If a structure contains some heavy atoms then some of the positions of the random atoms with highest CC-all may close to one of heavy atom positions. That will give a good enough initial phases, for example better than 80 degrees, to be refined by ACORN-PHASE to solve the structure.
Another procedure is a molecular replacement method of searching all possible sets of orientations and positions for the starting fragment. ACORN-MR will do the rotation function first and then translational function on the best solution of rotation function. The program will calculate CC-all for each set. Then all sets will be sorted on CC-all and the maximum 1000 sets of the initial phases and weights will be calculate with highest CC-all for ACORN-PHASE to refine. There are two kind of searching approaches: step by step searching and random searching. Normally random searching needs less computing time.
There are three procedures used for the phase refinement in ACORN-PHASE: Dynamic Density Modification (DDM), real space Sayre Equation Refinement (SER) and Patterson superposition (SUPP). For a default running only DDM is employed to refine the initial phases. DDM will calculate a map using a set of initial phases and weights of the strong reflections and modify the map according to the ratio of map density (RO) over map standard deviation (SIGRO) and cut the top density at a level (CUTD) from 3*SIGRO to 15*SIGRO (default) according the number of cycles. A new set of structure factors will be obtained from the modified map by FFT and CC-medium will be calculated to check if a solution is reached. The DDM can modify not only E or F maps, but also Patterson or sharpened Patterson maps because the DDM will treat all maps as a map of ratio RO/SIGRO.
DDM-Curve:
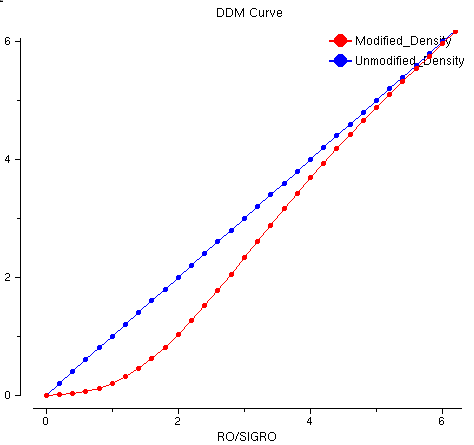
The DDM-Curve shows the approach of dynamic density modification that makes negative densities to zero and depresses the densities less than 1*SIGRO which mostly are noise and enhances the densities between 1*SIGRO and CUTD by cutting top densities in order to develop the new densities in protein region but outside the starting fragment region. Therefore the starting fragment including heavy atoms will not dominate the phase refinement process and the new densities in protein region will become more important.
In case of DDM alone did not reach the solution then SER should be introduced for a couple of cycles. SER uses a number of FFTs and inverse FFTs for all calculations to carry out the Sayre Equation refinement in real space. There are no phase relationships needed so that SER has no limit on the number of reflections to be used in the Sayre Equation. The purpose of using SER is to disturb the phase refinement process in DDM in order to reach global minimum other than local minimum.
SUPP calculates a half-sharpened Patterson and superposition the map by sum-function on the atom positions of the starting fragment. Then one cycle DDM will used to modify the map and a new set of phases will be calculated from the map. SUPP can be used only when the starting fragment is relatively large, say more than 10 atoms in it and normally the initial phase error can be reduce about 1 or 2 degrees.
A lot of protein structures contain Sulphur atoms which can be located using anomalous scattering differences. Since the anomalous signal from Sulphur atoms is weak it has to be careful to collect the anomalous scattering data. But if the size of the structure is between 100 and 200 amino acids the first thing to try is the use of ACORN with random atom searching to solve the structure with native atomic resolution data before trying to collect the the anomalous scattering data. If atoms which are heaver than Sulphur atoms are in the structure the random atom searching in ACORN can solve even big structures, such as the Sel-Met proteins where Sulphur atoms are replaced by Selenium atoms.
As other direct method programs ACORN with random atom searching can be used to locate anomalous scatterers from SAD or MAD data, even the data is at low resolution, say 3-4 Angstrom because the anomalous scatterers are far apart from each other and can still be resolved at such resolution. Then the positions of the anomalous scatterers can be input to ACORN to solve the complete structure with atomic resolution data. Any known positions of anomalous scatterers or heavy atoms located by other methods can be used by ACORN directly.
A large part of protein structures contains alpha helices which have very similar configurations. Therefore a standard alpha helices in CCP4 fragment library is a very good starting fragment for ACORN to solve such protein structures. ACORN can pick up a part of the standard alpha helices with the size to suit the structure to be determined, for example 50 atoms which are 10 Alanines. The random searching MR approach in ACORN-MR is advised to obtain a correct orientation and position quickly. Another way to obtain a starting fragment is searching Protein Data Bank (PDB) or other data bank to find a small motif. For example the sequence searching by netblast can be used because the sequence of the structure is normally known. ACORN-MR will carry out the same procedure to find the correct orientation and position based on highest CC-all.
If a large motif can be found for the structure to be determined AMORE can be used alternately to obtain the correct orientation and position and then ACORN can be used to refine the initial phases from the positioned motif. But the size of the motif has to be large enough for AMORE to find the correct orientation and position.
A small molecular structure can be easily solved by random atom searching in ACORN even the structure contains only light (C, N and O) atoms since the data normally has higher than 1.0 Angstrom resolution.
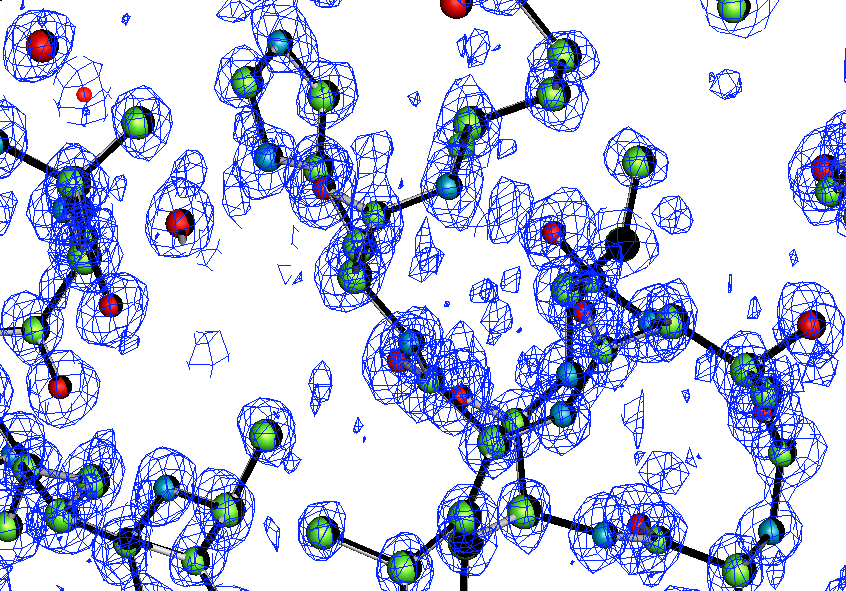
The following E-amp was calculated using the phases and weights from ACORN starting from Zinc position with 1eb6 atomic resolution data. It was quick to build the structure by ARP/wARP. The contour level of the E-map was 2*SIGRO. It is clear to see the E-map is very close to the final model.
E-map for 1eb6: