By A. Urzhumtsev
Laboratory of Crystallography and Modeling of Mineral and Biological Materials
University Henri Poincaré, Nancy 1, Vandoeuvre-les-Nancy, France
One of the main goals of structural crystallography is, given a crystal, to propose a model which will be chemically consistent and which will explain experimentally measured structure factor magnitudes. Currently, atomic models of spherical (isotropic) or elliptical (anisotropic) individual atoms are accepted as good at conventional resolutions, when the high resolution limit of the data set vary between roughly 1 and 5 Å. When the resolution of a data set is higher than 1 Å, more complicated models such as multipolar ones (Hansen & Coppens, 1978; Lecomte, 1998) are necessary. At the opposite end, at resolutions 5-10 Å and lower, atomic models are useless because they have too many parameters with respect to a very limited number of experimentally measured magnitudes. At the same time, at such resolutions there is another, more important problem. Starting roughly at 7-8 Å, these models being used alone do not explain diffraction data (see, for example, one of the pioneering works by Phillips, 1980).
A reason for such behaviour is the incompleteness of atomic models which miss the contribution of the bulk solvent :
This resolution limit of 7-8 Å shows that the solvent features are of this size (therefore they are not important at higher resolutions) and that in order to use corresponding diffraction data a model for the bulk solvent should be introduced. A review of possible approaches for such modelling is proposed below; it follows the talk given at the IUCr-18 (Urzhumtsev, 1999). A part of this presentation was inspired by the comparative analyses done by Jiang & Brünger (1994) and by Kostrewa (1997). The current review differs from the former by more schematised presentation and from the latter by including larger material. Other known reviews on the solvent modelling are those by Tronrud (1997) and Badger (1997).
Currently, macromolecular crystallographers use solvent modelling in two extreme cases : for well ordered (crystallographic) solvent molecules which are modelled exactly in the same way as the macromolecule itself and for completely disordered molecules which are modelled by a continuum function. These models are used mainly during atomic model refinement. An intermediate case of partially ordered solvent molecules can occur as it is the case for water channels. In this case also, as for "crystallographic" molecules, an atomic model can be produced (Podjarny et al., 1997) similarly to multicopy models of flexible chains (Burling & Brünger, 1994). On contrary, such (or other) models for the case of the bulk solvent are not yet developed and some approaches to do so are analysed below. It is important to note that this modelling is important at any stage of crystallographic studies when molecular models appear and not only for a refinement of atomic models.
Different approaches are possible to deal with the problem of the contribution of the bulk solvent to diffraction data or to corresponding crystallographic image:
The approaches 1-3 and 5 are discussed briefly in this Section, and the rest of the paper deals with different explicit models of the bulk solvent.
Exclusion of low resolution data
Until 90ths most of refinements were done excluding reflections of the resolution lower than 6-7 Å. This solved also another, technical problem of low resolution : measuring these reflections. Later an importance of low resolution data for the refinement (see, for example, Kostrewa, 1997) and for the map quality (Urzhumtsev, 1991) became accepted by the crystallographic community and currently this approach is no longer used. However, the same idea is exploited now for molecular replacement where the lower resolution limit used is about 10-15 Å. While such exclusion is not important (and seems to be even useful) for the rotation search, low resolution data can be extremely useful for the solution of the translation problem as it is shown by Urzhumtsev & Podjarny (1995a). A search with low resolution data is less sensitive to eventual orientation errors in the model. Even more, the possibility of low resolution direct phasing (see, for example, Lunin et al., 2000) can give a complementary way to solve the translation and in some case, the rotation problem.
Solvent flattening or relevant techniques
A known information on the density distribution in the solvent region can be used in order to improve available electron density maps. In most of cases, such distribution is supposed to be flat, and a number of solvent flattening techniques exist now being originated in its modern form by Bricogne (1974). This information can be used alone or together with other informations, for example, with a known histogram for the electron density distribution inside the molecular envelope (Zhang & Main, 1990).
It is important to remind that if the "observed" phases 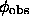 would be available the map calculated with them will not have a flat density in the solvent regions because of a limited resolution (Fourier truncation effects) and a usual absence of some reflections, specially those of very low resolution. In other words, flattening procedures look for some wrong phases which nevertheless produce a more interpretable image. Such effect was analysed by Brazhnikov et al. (1993) when the same quality of an improved map was obtained by a simple reconstruction and addition to the synthesis of few low resolution reflections or by iterative application of the solvent flattening procedure (Wang, 1985).
would be available the map calculated with them will not have a flat density in the solvent regions because of a limited resolution (Fourier truncation effects) and a usual absence of some reflections, specially those of very low resolution. In other words, flattening procedures look for some wrong phases which nevertheless produce a more interpretable image. Such effect was analysed by Brazhnikov et al. (1993) when the same quality of an improved map was obtained by a simple reconstruction and addition to the synthesis of few low resolution reflections or by iterative application of the solvent flattening procedure (Wang, 1985).
In fact, the most interpretable macromolecular map would be calculated rather with the coefficients
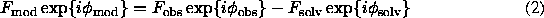
and not with

where the phases 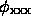 are found by some phase improvement technique. Naturally, in practice when the phases are unknown, the best known their approximation
are found by some phase improvement technique. Naturally, in practice when the phases are unknown, the best known their approximation 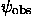 should be used:
should be used:
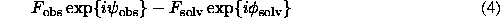
Maps with the coefficients (4) could be used before an atomic model is known in order to facilitate its construction and the map interpretation. Therefore, the first problem of the solvent modelling is:
| Problem 1: | how to estimate the bulk solvent contribution before an atomic model is known? |
| The goal here is to improve a map in order to build an atomic model of the molecule | |
Statistical approach
The equality (1) can be treated in a such way that a complete model of a crystal consists of a macromolecular model plus a number of atoms, ordered or not, which are not yet identified and not yet taken into account:
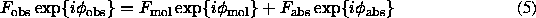
Under some statistical hypotheses on possible distribution of these absent atoms, the mean value for correcting structure factors 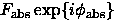 can be estimated and therefore the best macromolecular model (the most probable hypothesis on values of atomic parameters) can be chosen using maximum likelihood approach (Bricogne & Irwin, 1996, Pannu & Read, 1996; Read, 1997; Murshudov et al., 1997; Pannu et al., 1998; some comments on this subject can be found in Lunin & Urzhumtsev, 1999). One can accept a hypothesis that the missed atoms are, at the first approximation, randomly and uniformly distributed in the unit cell or another, better hypothesis, for example that they are distributed only in the solvent region, and, maybe, not uniformly. In any case, when refining an atomic model with the maximum likelihood approach, model structure factors Fmod are fitted to the values
can be estimated and therefore the best macromolecular model (the most probable hypothesis on values of atomic parameters) can be chosen using maximum likelihood approach (Bricogne & Irwin, 1996, Pannu & Read, 1996; Read, 1997; Murshudov et al., 1997; Pannu et al., 1998; some comments on this subject can be found in Lunin & Urzhumtsev, 1999). One can accept a hypothesis that the missed atoms are, at the first approximation, randomly and uniformly distributed in the unit cell or another, better hypothesis, for example that they are distributed only in the solvent region, and, maybe, not uniformly. In any case, when refining an atomic model with the maximum likelihood approach, model structure factors Fmod are fitted to the values
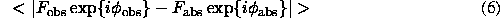
averaged over all possible positions of missed atoms (in the current study, supposed to be in the solvent region) and no longer to the experimental values 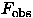 .
.
In such approach the knowledge of an approximate atomic model for the macromolecule is crucial and the problem differs from the Problem 1 discussed above:
Problem 2: how to estimate the bulk solvent contribution when an atomic model is known? | |
| The goal here is to obtain the best possible atomic model of the whole crystal which can be used in order to answer different physical, chemical or biological questions. | |
Phasing methods based on specific solvent diffraction
In contrast to previous, "passive" treatment of the solvent, an "active" way of doing this is possible where a variation of the solvent density allows to get phase values (contrast variation methods; Bragg & Perutz, 1952; Roth, 1987, 1991; Carter et al., 1990). Alternatively, the phases can be obtained from the same solvent but diffracting anomalously (MASK method by Fourme et al., 1994). These methods need to be analysed independently and will not be discussed here.
Molecular and solvent regions
The most direct way, supplying with most interesting information but not easy to realise is to build an explicit model of solvent. There are several suggested approaches discussed below.
In general, a model of the bulk solvent can be built both in real and in reciprocal space. In reciprocal space, it is a set of solvent structure factors, usually with the same indices as for the set of experimentally measured data while eventually it can be more complete or go to slightly higher resolution. In real space, a solvent model is usually a density distribution. This distribution can be either ideal or approximate at a given resolution.
In order to build a bulk solvent model in real space, one needs to define two objects :
a) solvent region and
b) density distribution inside this region.
Solvent region usually is considered as a part of the unit cell complementary to the molecular region (or molecular mask). Sometimes, a thin shell between them is initially excluded from both and then filled with more complicated procedures (Jiang & Brünger, 1994). Traditionally, the molecular region is represented by a binary function M(r) calculated in a chosen grid. Correspondingly to the two problems formulated in the previous section, two different situations are possible when an atomic model is known or not.
In the first case, the molecular mask M(r) is traditionally defined as a conjunction of spheres of a given radius centred in the atomic positions. This mask is final, exact (within the limits of the atomic positional errors, their radius etc).
In the second case, the molecular mask can be defined from an electron density distribution 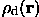 calculated at the resolution d. Molecular (solvent) region defined on the base of a density distribution does not have any longer an absolute meaning as M(r). Since all details with the resolution higher than d is absent in the map, the best mask image also cannot have high resolution details. The best possible envelope Md(r) at the resolution d can be imagined, for example, as a result of calculation of structure factors from M(r), suppression of high resolution reflections, calculation of a new function with the rest of structure factors and, finally, of a selection of points with highest values of this new Fourier synthesis.
calculated at the resolution d. Molecular (solvent) region defined on the base of a density distribution does not have any longer an absolute meaning as M(r). Since all details with the resolution higher than d is absent in the map, the best mask image also cannot have high resolution details. The best possible envelope Md(r) at the resolution d can be imagined, for example, as a result of calculation of structure factors from M(r), suppression of high resolution reflections, calculation of a new function with the rest of structure factors and, finally, of a selection of points with highest values of this new Fourier synthesis.
Naturally, when neither molecular mask M(r) nor an atomic models are known, the ideal Md(r) cannot be calculated and an approximation md(r) to it can be defined as a set of (grid) points with
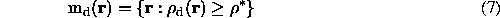
where 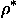 is chosen such that the volume of the region md(r) is roughly equal to the molecular volume. Clearly, the result strongly depends on the resolution and on the quality of the initial density
is chosen such that the volume of the region md(r) is roughly equal to the molecular volume. Clearly, the result strongly depends on the resolution and on the quality of the initial density 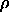 d(r) and, in particular, on errors in structure factors and on absence of some of them (Fig. 1). In many cases this procedure is not good because it gives a multidomain regions that does not agree with the idea to have a molecular mask as a single domain.
d(r) and, in particular, on errors in structure factors and on absence of some of them (Fig. 1). In many cases this procedure is not good because it gives a multidomain regions that does not agree with the idea to have a molecular mask as a single domain.
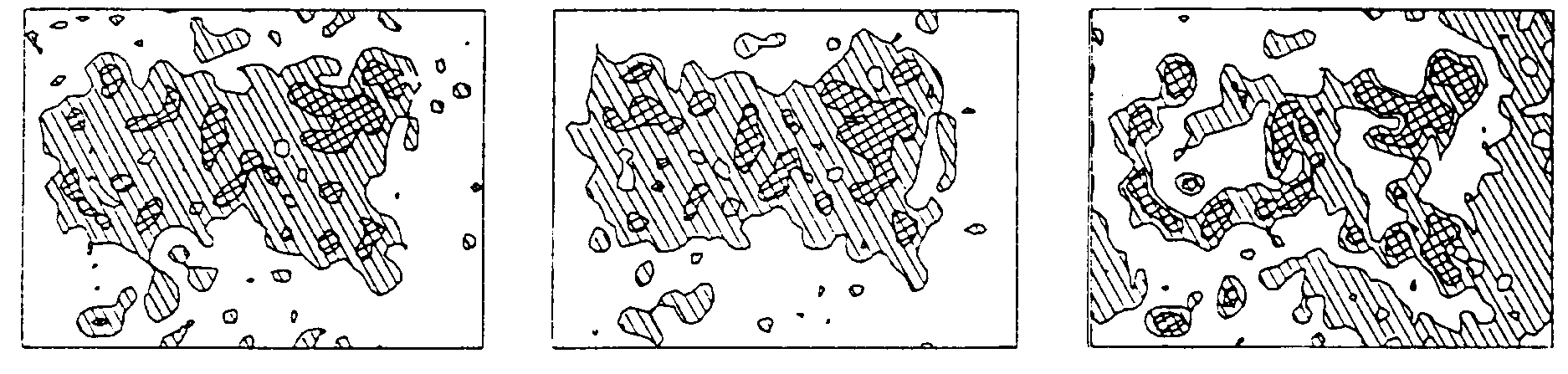
Fig. 1. Influence of the phase errors and the exclusion of low resolution data on the quality of molecular envelope; test model (Urzhumtsev, 1991). ideal map at 6Å resolution, about 2500 reflections used; external contour selects a half of the unit cell (left); SIR phased map with the same set of reflections (centre); the same structure factors as for the left figure except all 29 reflections with the resolution lower than 30Å excluded from the map calculation (right)
Map features and automatic mask determination
In order to determine the molecular mask from a density distribution, some key density features which allow to distinguish the macromolecular and solvent regions should be formulated. For every grid point where the density is calculated, these features should allow to make a choice whether the point belongs to the molecular region or not, or, in more soft way, with which probability it belongs to the molecular region. In other words, a density transformation

should be defined which for every point r assigns a probability value p(r) instead of the Fourier synthesis value d(r). In the simplest case (7) discussed above, this is a binary function
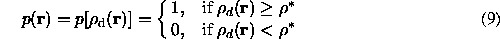
which depends on the value in a given point r. In the example of the density modification function suggested by Wang (1985) it is:
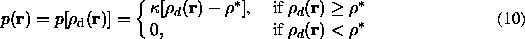
where 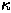 is a normalising factor. This step is logically executed in direct space and can be considered as a corresponding filtration procedure for .
is a normalising factor. This step is logically executed in direct space and can be considered as a corresponding filtration procedure for .
For noisy maps which is usually the case, such procedure is not sufficiently good and does not always give a single domain regions suggesting further treatment of this information. For example, for the binary selection (9), the molecular region can be considered as a zone of highest concentration of the selected points. If the resulted function p(r) is considered as a probability, then the molecular region can be defined as a zone where a whole sphere of a given radius likely belongs to the molecule. Both reasoning can be realised through a local averaging of the transformed function p(r) :
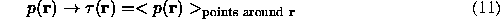
An understanding and implementation of this second step allowed to create an automated approach for envelope determination (Westbrook et al., 1984; Wang, 1985; Urzhumtsev, 1985; Jones et al., 1991). The result of averaging varies but not strongly with different weighting functions used during averaging; on contrary, the correct choice of the averaging radius is more important (Urzhumtsev, 1991). This step is easier to be done through structure factors calculation (Leslie, 1987; Lunin, in Urzhumtsev, 1985) and can be considered as a filtration procedure in reciprocal space.
Now, with a correct choice of the filtration parameters, a selection of highest probability points in 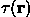 :
:
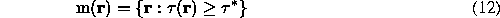
constructs a region, which consists of a single domain per molecule and has quite low probability of errors.
In spite of importance of the second step of averaging, it seems that it is the first step of density filtration (9-10) which plays the crucial role through the information used for the selection of points. Highest density values used by Wang (1985) and Westbrook et al. (1984) is one example of such information (Fig. 2). It was noted by Urzhumtsev et al. (1989) that due to the truncation effects the points with lowest density values at a synthesis of a finite resolution also indicate the molecular region (Fig. 2). A similar observation was used by Jones et al. (1991) who noted that the points inside the molecular region correspond to highest local density fluctuation; in this case the density filtration function depends not only on a density value in a given point like in (9) or (10) but on value in several points.
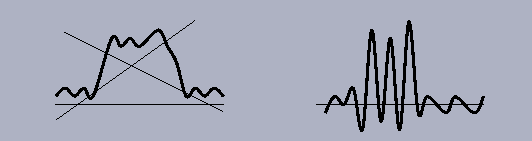
Fig. 2. Schematic one-dimensional representation of the distribution of the values of the Fourier synthesis calculated at a limited resolution. In many cases, molecular region contains not only highest density values but also lowest density values and has points with highest local density fluctuation.
For cases with a known non crystallographic symmetry, some automated procedures were suggested (Rees et al., 1990) based on the similarity of the density values for the symmetrically related points differentiating them from the points of solvent for which this symmetry is not applicable.
Flat envelope. Mask method.
When the solvent region is determined, the next step is to construct a density distribution inside this region. This problem was addressed several times from the beginning of 50ths (Wrinch, 1950; Bragg & Perutz, 1952; Langridge et al., 1960; Fraser et al., 1965, 1978; O'Brien & MacEwan, 1970) for several particular problems. Later Phillips (1980) published a clear evidence of a discrepancy between experimental and calculated structure factors magnitudes for a macromolecular case, indicated resolution limits for this and proposed a scheme of a correction for the bulk solvent which with some variations is used until nowadays:
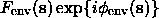 for this flat molecular mask; following the Babinet principle, structure factors for a flat solvent region are opposite to ;
for this flat molecular mask; following the Babinet principle, structure factors for a flat solvent region are opposite to ;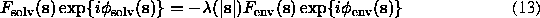
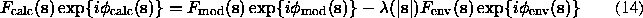
The basic hypothesis of this scheme is a flat density distribution in the solvent region. Here, a macromolecular atomic model is supposed to be known and 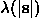 is a function, usually gaussian, of the resolution:
is a function, usually gaussian, of the resolution:
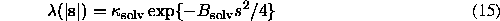
Its parameters (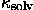 and Bsolv in this particular example) are chosen from the best fit of Fcalc(s) to Fobs(s) for a given fixed macromolecular model. This approximation may be not always efficient. An example is the case of aldose reductase (Rondeau et al., 1992) for which the mean value varies rather as a sigmoid and not as a gaussian function (Fig. 3). On the other hand, the approximation (15) is important only up to the resolution of about 5 Å above which the solvent contribution is negligible; in this sense, the case of aldose reductase does not really contradict the approximation. Another possibility for is a local scaling discussed, for example, by Tronrud (1997).
and Bsolv in this particular example) are chosen from the best fit of Fcalc(s) to Fobs(s) for a given fixed macromolecular model. This approximation may be not always efficient. An example is the case of aldose reductase (Rondeau et al., 1992) for which the mean value varies rather as a sigmoid and not as a gaussian function (Fig. 3). On the other hand, the approximation (15) is important only up to the resolution of about 5 Å above which the solvent contribution is negligible; in this sense, the case of aldose reductase does not really contradict the approximation. Another possibility for is a local scaling discussed, for example, by Tronrud (1997).
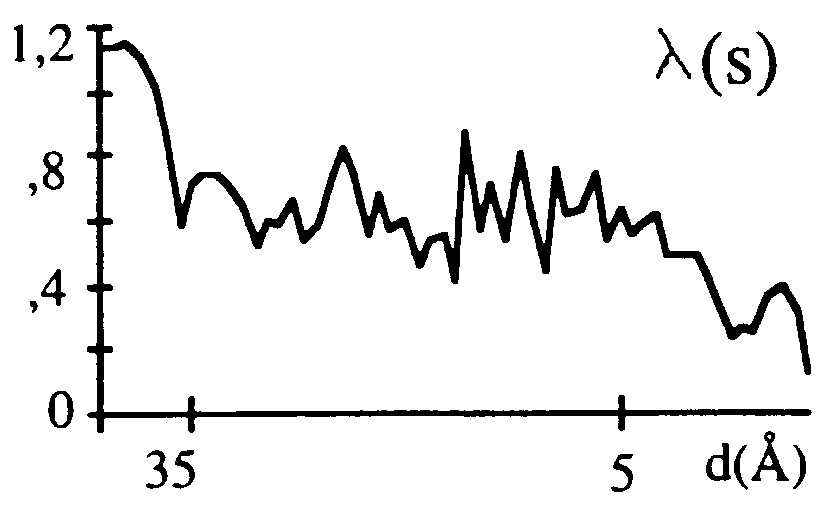
Fig.3. Optimal scaling coefficient for the envelope structure factors as a function of the resolution.
Among the variations introduced into the initial scheme, a special procedure to assign the density values for the points of a narrow shell between the molecular and solvent regions (Jiang & Brünger, 1994) is of high importance (Kostrewa, 1997).
While the method, realised for example through X-PLOR (Brünger, 1992) and CNS (Brünger et al., 1998), allows to reduce dramatically the discrepancy between observed and calculated low resolution structure factors magnitudes, a detailed analysis by Jiang & Brünger (1994) allowed them to conclude that "the best solvent models results in R-factors significantly higher than one might expect". Therefore, two questions arise :
Flat envelope. Exponential scaling model.
A simplification of the previously described method was suggested following the argumentation that the bulk solvent correction is important only at low resolution where density both in macromolecular and in solvent region can be considered as flat. In this case, structure factors calculated for two binary (flat) functions, complementary in the unit cell, are proportional each to other (see, for example, Langridge et al., 1960 or Tronrud, 1997):
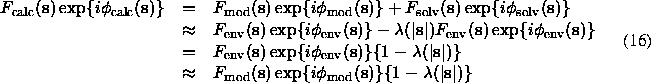
(Fmod(s) and Fenv(s) are supposed to be quite close).
Because the values usually vary between 0 and 1, this formula (16) illustrates the phenomenon that at low resolution observed structure factor magnitudes are lower than those calculated from the macromolecular model. The formula (16) suggests also two ways to diminish the discrepancy between mean values of magnitudes: either observed magnitudes Fobs(s) should be corrected as
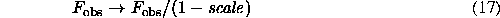
before being compared with Fmod(s) , or the model structure factors should be the multiplied by 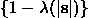 . The latter can be also done by correction of atomic scattering factors (Fraser et al., 1978).
. The latter can be also done by correction of atomic scattering factors (Fraser et al., 1978).
The key question in this approach is whether such linearity between molecular and envelope structure factors holds up to a reasonable resolution. Some tests were done with the diffraction data for aldose reductase (Rondeau et al., 1992). For this case of a well refined atomic model, solvent structure factors as complex numbers were estimated and compared with the molecular structure factors (Urzhumtsev & Podjarny, 1995b; Podjarny & Urzhumtsev, 1997). The results of such comparison (Fig. 4) can be interpreted so that these structure factors are approximately anti-collinear at the resolution lower than 12-15 Å, and are not correlated at higher resolutions. This shows that the scaling method has a limited range of application in comparison with the mask method what was found experimentally (Kostrewa, 1997).
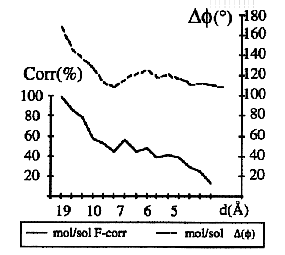
Fig.4. Comparison of model and solvent structure factors. Amplitude correlation and phase difference
Distance dependent solvent modelling
In order to improve the quality of solvent modelling obtained by the mask method, several efforts were undertaken. The first idea was to replace a flat solvent density by some more complicated distribution. As it is known, the solvent molecules are distributed in shells, and a logical step is to model the solvent density as a function dependent on the distance r to the molecular border (Schoenborn, 1988; Cheng & Schoenborn, 1990).
In this case, the knowledge of a molecular model is important in order to reproduce correctly the shells. While such idea is very attractive, an analysis done by Jiang & Brünger (1994) showed that the improvement obtained by this method is marginal in comparison with the mask method.
One of possible explanation is that in fact while the mean density in the shells correlates with the distance to the molecular border, the distribution of density in every shell is far from be uniform. In other words, the density in the points with the same distance to the molecular border is rather different and can depend on the shape of the border (cavity, channel, etc.; a nice illustration for this can be obtained in Levitt & Park, 1993). Such points can be distinguished if the molecular envelope Md(r) or its approximation md(r) is calculated at several different resolutions d (Fig. 5; see also Section 3 for Md(r) calculation). Small cavities, after the map is recalculated without higher resolution reflections, become hidden, and corresponding points outside the envelope become inside it.
An additional advantage of such approach is that eventually it does not need a molecular atomic model. If molecular envelopes in the crystal are known at the resolutions d and below, this could be eventually enough to reproduce the solvent density distribution at the same resolution d (or, maybe, slightly lower).
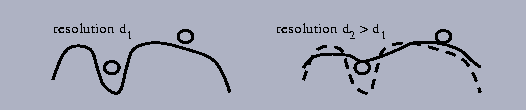
Fig. 5. Schematic presentation of the difference in the relative position of water molecules with respect to the molecular envelope calculated at different resolution.
2D density histograms
In order to use these ideas, some initial information on typical density distributions should be collected. This information can be obtained and reproduced using two-dimensional distance-dependent density histograms. If for a given crystal a molecular envelope can be calculated at several resolution dn, n = 1, ... , N, then for a density map D(r) calculated at the resolution D and for every such envelope Mn(r) a common distribution of (,r) can be calculated where r is the distance from a given point r to the molecular envelope and is the density value in this point. Par analogy with usual density histograms HD() (Lunin, 1988), these common distributions can be considered as two-dimensional histograms HD,d(,r).
This information can be used in the following way (Urzhumtsev & Podjarny, 1995b; Urzhumtsev et al., 2000).
If a single envelope Md(r) and the corresponding 2D-historgam HD,d(,r) are known, then for every point r in the unit cell, first, its distance r* to the envelope is calculated, second, a one-dimensional density distribution hD,d() = HD,d(,r*) corresponding to this distance is extracted from HD,d(,r) and, third, the density value in this point is assigned to be equal to the mean (or to the most probable) value of this distribution hD,d(). The procedure is essentially the same as the procedure suggested by Schoenborn (1988) with the difference that it does not need to know an atomic model and reconstructs not the ideal solvent density distribution but its image at the resolution D.
However, when several envelopes Mn(r) and histograms HD,n(,r) are known for the resolutions dn, n = 1, ... , N, then for every point r a set of one-dimensional distributions hD,n() = HD,n(,rn*) can be obtained that allows to identify better the position of this point with respect to the molecular surface as it is discussed above. These distributions can be multiplied (this is a very crude treatment of the information because naturally these distributions hD,n() are not independent) and, again, the mean (or the most probable) value of this product can be assigned as the density value in this point. It is easy to see that this approach can have larger applications and be used for the density reconstruction in the whole unit cell and not only for the solvent.
The first analysis done with the experimental data for aldose reductase (Rondeau et al., 1992) showed that indeed such approach allows reasonably well to reconstruct the density but the quality of solvent models, in spite of more detailed density function, is not better either that the result obtained by the "standard" mask approach.
Atomic modelling
An idea to build an atomic model for the bulk solvent is attractive as a logical continuation of a models for "crystallographic" and for partially ordered water molecules (Podjarny et al., 1997). Some variant was tested quite a long ago (O’Brien & MacEwan, 1970) when random atoms were placed inside the macromolecular envelope in order to calculate structure factors using the Babinet principle. However, a use of "bulk solvent atoms" together with the macromolecular atoms in general is not possible because it essentially increases the number of parameters and makes it too high with respect to the number of experimental data. Therefore, some more delicate approaches could be proposed in future.
Difference density approach
Several ideas to construct a density model are based on the analysis of the difference density which is not yet interpreted by the current macromolecular model. Badger & Caspar (1991) and Badger (1993) suggested to include iteratively the peaks in the solvent region of the difference map
into the solvent model and to suppress these peaks if necessary in following iterations. This idea reminds an approach for direct phasing described by Simonov (1976) with the difference that here the density is analysed only in the solvent region. An opposite, in certain sense, hypothesis that the large positive and negative peaks do not correspond to any features but exist due to important phase errors and should be removed from the map was used for solvent correction by Jiang & Brünger (1994). However, the same authors found both difference density approaches overfitting the model and giving quite marginal improvement in comparison with the mask method.
The mask approach is a simple method for solvent modelling which gives a reasonable estimation for bulk solvent contribution. However, this model is not yet sufficiently good and its application needs a knowledge of a macromolecular atomic model.
Currently, several problems of macromolecular crystallography are relevant to the problem of the bulk solvent modelling. First, the work at extra high resolutions with more structural details and studies of molecular potentials needs better solvent models. Second, a work with crystals of not sufficient quality and an interpretation of corresponding density maps could be advanced if the solvent contribution can be estimated and removed from the maps before an atomic model is known. Finally, a fast estimation of the solvent contribution could be useful in the search procedures where many different molecular positions must be checked like in molecular replacement. These problems of solvent modelling are open for further researches.
The author thank D.Kostrewa, V.Lunin and A.Podjarny for many useful discussions on the subject and critical discussion of the manuscript and C.Lecomte for his interest to this work.
Badger, J. (1993) Multiple hydration layers in cubic insulin crystals. Biophys. J. 65, 1656-1659.
Badger, J. (1997) Modeling and Refinement of Water Molecules and Disordered Solvent. In Methods in Enzymology, Academic Press, San Diego., C.W.Carter, Jr., R.M.Sweet, eds. 277B, 344-352
Badger, J. & Caspar, D.L.D. (1991) Water structure in cubic crystal. Proc.Natl.Acad.Sci.USA 88, 622-626.
Bragg, W.L. & Perutz, M.F. (1952). The External Form of the Haemoglobin Molecule. I. Acta Cryst. 5, 277-283.
Brazhnikov, E., Chirgadze, Yu., Aevarsson, A., Svensson, A., & Lunin, V. (1993). The Similarity of Results Obtained by Solvent Flattening and Low-Resolution Phase Retrieval for the Improvement of Protein Electron Density Map. 3rd European Workshop of Biological Macromolecules, Centro di Cultura Scientifica «A.Volta», Como (Italy), May 21-25, 1993. M4.
Bricogne, G. (1974) Geometric Sources of Redundancy in Intensity Data and Their Use for Phase Determination. Acta Cryst, A30, 395-405.
Bricogne, G. & Irwin, J. (1996) In Macromolecular Refinement: Proceedingof the CCP4 Study Weekend, E.Dodson, M.Moore, A.Ralph & S.Bailey, eds., pp.85-92. Warrington : Daresbury Laboratory.
Brünger, A. T. (1992) X-Plor, Version 3.1: A System for X-ray Crystallography and NMR. Yale University Press, New Haven, CT
Brünger, A.T., Adams, P.D., Clore, G.M., DeLabo, W.L., Gros, P., Grosse-Kunstleve, R.W., Jiang, J.-S., Kuszewski, J., Nilges, M., Pannu, N.S., Read, R.J., Rice, L.M., Simonson, T. & Warren, G.L. (1998) Crystallography & NMR System: A new Software Suite for Macromolecular Structure Determination. Acta Cryst., D54, 905-921.
Burling, F.T. & Brünger, A.T. (1994) Thermal motion and conformational disorder in protein crystals structures: Comparison of multi-conformer and time-averaging models. Isr.J.Chem., 34, 165-175.
Carter C.W., Jr., Crumbley, K.V., Coleman, D.E., Hage, F. & Bricogne, G. (1990) Direct Phase Determination for the Molecular Envelope of Tryptophanyl-tRNA Synthetase from Bacillus stearothermophilus by X-ray Contrast Variation. Acta Cryst. A46, 57-68.
Cheng, X.D. & Schoenborn, B.P. (1990) Hydration is protein crystals. A neutron diffrcation analysis of carbonmonoxymyoglobin. Acta Cryst., B46, 195-208
Fraser, R.D.B., MacRae, T.P. & Miller, A. (1965) X-Ray diffrcation Patterns of
a-Fibrous Proteins. J.Mol.Biol., 14, 432-442Fraser, R.D.B., MacRae, T.P & Suzuki, E. (1978) An Improved Method for Calculating the Contribution of Solvent to the X-ray Diffraction Pattern of Biological Molecules. J.Appl.Cryst., 11, 693 -694
Fourme, R., Shepard, W., Hermite, G.L. & Kahn, R. (1994) The Multiwavelength Anomalous Solvent Contrast Method (MASC) in Macromolecular CrystallographyIn "ACA Annual Meeting, June 25 - July 1, 1994", p.40. INFORUM, Atlanta, Georgia, Atlanta Convention Center..
Hansen, N.K. & Coppens, P. (1978) Testing Aspherical Atom Refinement on Small-Molecule Data Sets. Acta Cryst., A34, 909-921.
Jiang, J.-S. & Brünger, A.T. (1994) Protein Hydration Observed by X-ray Diffrcation. Solvation Properties of Penicillopepsin and Neuraminidase Crystal Structures. J.Mol.Biol., 243, 100-115
Jones, E.Y., Walker, N.P. & Stuart, D.I. (1991) Methodology Employed for the Structure Determination of Tumour Necrosis Factor, a Case of High Non-Crystallographic Symmetry. Acta Cryst. A47, 753-770.
Kostrewa, D. (1997) Bulk Solvent Correction : Practical Application and Effects in Reciprocal and Real Space. Joint CCP4 and ESF-EACBM Newsletter on Protein Crystallography, 34, 9-22.
Langridge, R., Marvin, D.A., Seeds, W.E., Wilson, H.R., Hooper, C.W., Wilkins, M.H.F. & Hamilton, L.D. (1960) The Molecular Configuration of Deoxyribonucleic Acid II. Molecular Models and their Fourier Transforms J.Mol.Biol., 2, 38-64
Lecomte, C. (1998) NATO ASI and Euroconference, NATO ASI Book, Kluwer Acad.Pub.Netherlands
Leslie, A.G.V. (1987) A reciprocal-space method for calculating a molecular envelope using the algorithm of B.C.Wang. Acta Cryst. A43, 134-136.
Levitt, M. & Park, B.P. (1993) Water: now you see it, now you don’t. Structure, 1, 223-226
Lunin, V.Yu. (1988) Use of the Information on Electron Density Distribution in Macromolecules. Acta Cryst. A44, 144-150.
Lunin, V.Y. & Urzhumtsev, A.G. (1999) Maximal Likelihood refinement. It works, but why ? CCP4 Newsletter on Protein Crystallography, 35, 8, 13-28
Lunin, V.Yu., Podjarny, A. & Urzhumtsev, A.G. (2000) Low resolution phasing : advances and perspectives. Acta Cryst., D56, sent to editor
Murshudov, G.N., Vagin, A.A. & Dodson, E.J. (1997) Refinement of Macromolecular Structures by the Maximum-Likelihood Method. Acta Cryst. D53, 240-255.
O’Brien, E.J. & MacEwan, A.W. (1970) Molecular and Crystal Structure of the Polynucleotide Com)plex: Polynosinic Acid plus Polydeoxycydylic Acid. J.Mol.Biol., 48, 243-261
Pannu, N.S. & Read, R.J. (1996) Improved Structure Refinement Through Maximum Likelihood. Acta Cryst. A52, 659-668.
Pannu, N.S., Murshudov, G.N., Dodson, E.J. & Read, R.J. (1998) Incorporation of Prior Phase Information Strengthens Maximum-Likelihood Structure Refinement. Acta Cryst. D54, 1285-1294
Phillips, S.E.V. (1980) Structure and Refinement of Oxymyoglobin at 1.6Å Resolution. J.Mol.Biol., 142, 531-554
Podjarny, A.D. & Urzhumtsev, A.G. (1997) Low resolution phasing. In Methods in Enzymology, Academic Press, San Diego., C.W.Carter, Jr., R.M.Sweet, eds. 276A, 641-658
Podjarny, A.D., Howard, E.I., Urzhumtsev, A.G. & Grigera, J.R. (1997) A multicopy modeling of the water distribution in macromolecular crystals; towards the compatibility between diffraction and NMR data. Proteins, 28, 303-312
Read, R.J. (1997) Model phases : Probabilities and Bias. In Methods in Enzymology, Academic Press, San Diego., C.W.Carter, Jr., R.M.Sweet, eds., 277B, 110-128.
Rees, B., Bilwes, A., Samama, J.P. & Moras, D. (1990) Cardiotoxin V''4 form Naja mosambica mosambica. The refined crystal structure. J.Mol.Biol., 214, 281-297
Roth, M. (1987) Best Density Maps in Low-Resolution Crystallography with Contrast Variation. Acta Cryst. A43, 780-787.
Roth, M. (1991) Phasing at Low Resolution. In "Crystallographic Computing 5" , Moras, D., Podjarny, A.D. and Thierry, J.-C., eds., p.229-248. Oxford University press.
Rondeau, J.-M., Tete-Favier, F., Podjarny, A., Reymann, J.-M., Barth, P., Biellmann, J.-F. & Moras, D. (1992) Novel NADPH-binding domain revealed by the crystal structure of aldose reductase. Nature, 355, 469-472.
Schoenborn, B.P. (1988) Solvent effect in protein crystals. A neutron diffraction analysis of solvent and ion density. J.Mol.Biol., 201, 741
Simonov, V.I. (1976) Phase refinement by the method of modification and Fourier transformation of an approximate electron density distributions. In : Crystallographic Computing Techniques, Ahmed, F.R., Huml, K., Sedlacek, B., eds. Copenhagen : Munskgaard, 138-143
Tronrud D. (1997) TNT Refinement Package. In Methods in Enzymology, Academic Press, San Diego., C.W.Carter, Jr., R.M.Sweet, eds., 277B, 306-319.
Urzhumtsev, A.G. (1985). The Use of Local Averaging to Analyze Macromolecular Images in the Electron Density Maps. ONTI NCBI, USSR Acad. of Sci., Pushchino.
Urzhumtsev, A.G. (1991) Low-Resolution Phases: Influence on SIR Syntheses and Retrieval with Double-Step Filtration. Acta Cryst., A47, 794-801.
Urzhumtsev, A.G. (1999) Solvent modelling at low resolution. Collected Abstracts, XVIIIth IUCr Congress & General Assembly, 4-13 August 1999, Glasgow, Scotland, M09.AA.003, 122
Urzhumtsev, A.G., Lunin, V.Yu. & Luzyanina, T.B. (1989) Bounding a Molecule in a Noisy Synthesis. Acta Cryst., A45, 34-39.
Urzhumtsev, A.G. & Podjarny, A.D. (1995a) On the Solution of the Molecular-Replacement Problem at Very Low Resolution: Application to Large Complexes. Acta Cryst, D51, 888-895.
Urzhumtsev, A.G. & Podjarny, A.D. (1995b) On the problem of solvent modeling in macromolecular crystals using diffraction data: 1. The low-resolution range. Joint CCP4 and ESF-EACBM Newsletter on Protein Crystallography, 32, 12-16.
Urzhumtsev, A.G., Podjarny, A., Lunin, V.Yu. (2000) Density information and low resolution phasing. Acta Cryst., D56, sent to editor
Wrinch, D. (1950) Vector maps of hydrated protein crystals. Acta Cryst., 3, 475-476
Wang, B.C. (1985) Resolution of phase ambiguity Methods Enzymol., 115, 90-112.
Westbrook, E.W., Piro, O.E. & Sigler, P.B. (1984) The 6-Å crystal structure of delta5-3-ketosteroid isomerase J.Biol.Chem. 259, 9096-9103.
Zhang, K.Y.J. & Main, P. (1990) Histogram Mathing as a New Density Modification Technique for Phase Refinement and Extension of Protein Molecules. Acta Cryst. A46, 41-46.