The CCP4 program suite contains two programs for calculating the solvent accessible surface area (ASA) of macromolecules: SURFACE and AREAIMOL. The two programs use different algorithms for the area calculations and offer different ranges of funtionality. In particular, AREAIMOL has recently undergone a number of changes which have enhanced its functionality and usability.
This article examines and compares the two programs.
The concept of the solvent accessible surface of a protein molecule was originally introduced by Lee and Richards (1971), as a way of quantifying hydrophobic burial. The solvent accessible area (ASA) describes the area over which contact between protein and solvent can occur.
| |
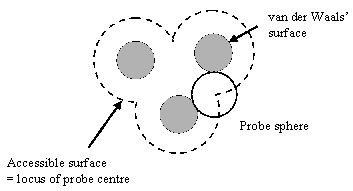
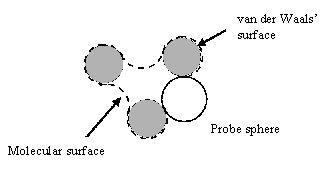
| Figure 1: accessible surface of a molecule, defined as the locus of the centre of a solvent molecule as it rolls over the Van der Waals surface of the protein. | Figure 2: molecular surface of a molecule, defined as the locus of the inward-facing probe sphere. |
The solvent accessible surface is defined as the locus of the centre of a probe sphere (representing the solvent molecule) as it rolls over the Van der Waals surface of the protein (illustrated for a simple case in figure 1). It is important to realise that this is different to the molecular surface, which is defined as the locus of the inward-facing probe sphere (Richards 1977) (illustrated in figure 2).
The original motivation for the calculation of the accessible surface was the study of the protein-folding problem and hydrophobicity. The size of the solvent accessible surface area buried in an interaction between protein units can be used to discriminate between crystal packing and a functional protein-protein interaction (used for example in the EBI's Quaternary Structure File Server (PQS), see http://pqs.ebi.ac.uk/pqs-doc/pqs-doc.shtml).
There are two main differences between AREAIMOL and SURFACE which can impact on the estimates of ASA which they produce. The first is that they use different algorithms to calculate ASA; this is examined in section 3.1 below. The second difference is the choice of van der Waals radii assigned to non-hydrogen atoms in the calculations, and this is examined in section 3.2.
Other differences are more to do with the ranges of functionality offered by the programs, and are discussed briefly in section 3.3. These differences shouldn't have an effect on the ASA estimates.
Finally, it is worth mentioning a couple of the similarities between the two programs. Both programs have parameters which control the precision of the area calculation (ZSTEP in SURFACE, PNTDEN in AREAIMOL). In either case, higher precision means relatively longer running times (though this is not a significant overhead in practice). Both programs also have a PROBE keyword, used to define the radius of the probe (solvent) molecule. It is usual to assume that accessibility to water is being assessed, and so in these tests the standard radius of 1.4 Å was used (the default for both programs).
In all cases the solvent accessible surface can be obtained by drawing a sphere around each atomic position with a radius equal to the van der Waals' radius of the atom plus the radius of the probe sphere (referred to as the expanded atom radius). The union of the expanded atoms is then the solvent-excluded volume.
The original Lee and Richards alogorithm for calculating ASA, implemented in the program SURFACE, effectively "slices" the expanded atom volume into 2-dimensional cross-sections along a single direction. Each slice contributes a surface area equal to the perimeter of the cross-section multiplied by the spacing between the slices. In SURFACE the keyword ZSTEP controls the fineness of the slices (values vary between 0.1 for most accurate, to 0.5 being the coarsest - default is 0.25).
The AREAIMOL program uses a different method, after Shrake and Rupley (1973). Dots are placed on the expanded atom surfaces, with a fixed number of dots per unit area. Dots which are not inside any other expanded sphere are considered to be accessible to solvent. The number of accessible dots multiplied by the density of the dots gives the accessible area on each atom. The keyword PNTDEN defines the number of points generated per square Angstrom on the surface (values go from the default of 1 to 100).
A number of simple tests were performed to assess the algorithms and compare their performance, as implemented in the two programs.
The values obtained from the programs for different values of ZSTEP/PNTDEN are given in tables 1 and 2, and in each case are compared with the analytical values, which can be calculated directly.
| SURFACE | AREAIMOL | ||
| ZSTEP | ASA | PNTDEN | ASA |
| 0.5 | 116.9 | 1 | 117.0 |
| 0.25 | 121.7 | 10 | 116.9 |
| 0.1 | 120.8 | 100 | 116.89 |
| Theoretical | 120.76 | Theoretical | 116.90 |
| SURFACE | AREAIMOL | ||||
| ZSTEP | A1 | A2 | PNTDEN | A1 | A2 |
| 0.5 | 90.5 | 82.9 | 1 | 89.0 | 86.0 |
| 0.25 | 95.9 | 76.9 | 10 | 89.8 | 85.1 |
| 0.1 | 94.7 | 78.0 | 100 | 89.46 | 85.29 |
| Theoretical | 94.73 | 78.03 | Theoretical | 89.51 | 85.29 |
| SURFACE | AREAIMOL | |||
| ZSTEP | ASA | PNTDEN | ASA | |
| a) VAL2 residue in RNAse Sa | 0.5 | 265.5 | 1 | 261.0 |
| 0.25 | 264.3 | 10 | 264.5 | |
| 0.1 | 265.0 | 100 | 264.8 | |
| b) SER3 residue in RNAse Sa | 0.5 | 233.5 | 1 | 236.0 |
| 0.25 | 231.9 | 10 | 233.3 | |
| 0.1 | 234.5 | 100 | 234.5 | |
| c) chain "A" in RNAse Sa | 0.5 | 5593.0 | 1 | 5587.0 |
| 0.25 | 5593.6 | 10 | 5565.5 | |
| 0.1 | 5571.6 | 100 | 5570.2 | |
Comments and Analysis
There seems to be a generally held opinion that ``surface dot'' methods such as that used in AREAIMOL are somehow inherently less accurate than those using the original Lee and Richards method (see for example Connolly (1996)).
This is not supported by these results. In the case of single/pairs of atoms, the ASA estimates from the two programs (obtained using the highest values of ZSTEP/PNTDEN) differ from the appropriate theoretical values by less than 0.1% in each case. In the larger examples the estimates of the ASA from SURFACE and AREAIMOL also differ from each other by less than 0.1%.
In fact this is much smaller than the variation due to using different values of PNTDEN/ZSTEP - in the worst cases, around 3-6% for different values of ZSTEP in SURFACE and 1.4% for different values of PNTDEN in AREAIMOL.
Finally, it is interesting to examine the extra time associated with running the programs using higher values for ZSTEP/PNTDEN. Table 4 gives a summary of the elapsed times for SURFACE and AREAIMOL when calculating ASA for chain A of RNAse Sa:
| SURFACE | AREAIMOL | ||
|---|---|---|---|
| ZSTEP | Elapsed time (s) | PNTDEN | Elapsed time (s) |
| 0.5 | 4.0 | 1 | 0.5 |
| 0.25 | 7.2 | 10 | 1.4 |
| 0.1 | 17.2 | 100 | 11.0 |
Using the highest values of ZSTEP/PNTDEN significantly increases the running time relative to using lower values - but compared to the time taken to run (for example) a refinement job, these times are negligible. It would certainly be realistic to use the programs with ZSTEP=0.1/PNTDEN=100 as the defaults.
Hydrogen atoms are not considered individually in the calculations (because it is not usual for them to be included in coordinate files describing the protein atom positions). The van der Waals' radii used for non-hydrogen atoms are therefore modified to account for the implicit presence of hydrogens. The choice of van der Waals' radii used in the calculations would also be expected to have an effect on the estimates of ASA.
SURFACE has two sets of radii taken from the literature: the first from Lee and Richards' original paper, and the second from Chothia (1975). The values are summarised in the table below:
| Lee and Richards | Chothia | |||
|---|---|---|---|---|
| Main-chain alpha carbon | 1.70 Å | Oxygen | 1.40 Å | |
| Main-chain carbonyl oxygen | 1.52 Å | Trigonal nitrogen | 1.65 Å | |
| Main-chain amide NH group | 1.55 Å | Tetrahedral nitrogen | 1.50 Å | |
| Main-chain carbonyl carbon | 1.80 Å | Tetrahedral carbon | 1.87 Å | |
| All side-chain atoms and groups | 1.80 Å | Trigonal carbon | 1.76 Å | |
| Sulphur | 1.85 Å | |||
| Water | 1.40 Å | |||
The Lee and Richards values are the default in SURFACE, and are selected using the VDWR RICH keywords; Chothia's values are selected using the VDWR CHC keywords. (AREAIMOL uses a simpler scheme where the same van der Waals' radius is assigned to all carbons, all oxygens and all nitrogens, regardless of the number of associated hydrogens.)
To investigate the effects of using different sets of van der Waals' radii, ASA was calculated using SURFACE with either VDWR RICH or VDWR CHC, for the residues and molecule used in the previous examples. The results are given in table 5, along with previous calculations using the same set of radii as AREAIMOL. ZSTEP was set to 0.1 in all the calculations.
| a) VAL2 residue | b) SER3 residue | c) Chain A | |
| SURFACE with VDWR RICH | 272.8 | 233.7 | 5591.6 |
| SURFACE with VDWR CHC | 263.0 | 225.7 | 5570.2 |
| SURFACE with values from AREAIMOL | 265.0 | 234.5 | 5571.6 |
| AREAIMOL | 264.8 | 234.5 | 5570.2 |
Comments and Analysis
In the examples considered, the percentage differences in ASA due to using different sets of van der Waals radii seem to decrease with increasing numbers of atoms considered - for the two residues considered the range of values differed by 3-4%, for the single molecule they differed by less than 1%. Interestingly, these are smaller variations than those due to changing ZSTEP (see the results in section 3.1).
An effect of different van der Waals' radii which was not considered here was the impact on the ASA of an individual atoms within a residue or molecule. An atom with a small ASA under one scheme might have zero ASA under the other (i.e. being buried completely by its neighbours). Whether this is important depends very much on the situation under investigation.
Ultimately the choice of program to use will be most likely be dictated by the problem the user is looking at.
SURFACE offers a powerful method for selecting which atoms are included in the calculations. The user is able to specify which atoms are to have their area calculated, which are to be included (so that they can ``obscure'' accessible area on other atoms, without their own area being considered), and which are to be excluded entirely (and thus play no part at all in the calculations). The option to use different sets of van der Waals radii has already been discussed in section 3.2 above. The output of SURFACE is used to prepare input for the VOLUME program.
On the downside, SURFACE (or rather, its users) suffers from an anachronistic input procedure and the program output is somewhat limited. It is also unable to directly examine crystallographic contacts or area differences.
AREAIMOL offers a very different range of functions. Most importantly, it is able to examine the effects of intermolecular and crystallographic contacts on the ASA, and can be used to look at ASA differences directly in a number of different situtations. It also offers a fairly comprehensive analysis of the ASA values, breaking them down by residue, chain and molecule. However there are equivalents to SURFACE's atom selection options, and a much simpler assignment of van der Waals radii is employed.
The functions offered by each program are summarised in table 6.
| Function | SURFACE | AREAIMOL |
|---|---|---|
| Area on individual atoms? | yes | yes |
| Atom selection? | yes | no |
| Different VDWR radii? | yes | no |
| Intermolecular/crystallographic contacts? | no | yes |
| ASA differences? | no | yes |
| Analysis of output? | no | yes |
| Prepares input for VOLUME? | yes | no |
A small number of tests have been performed to examine the estimates of ASA obtained from the CCP4 programs AREAIMOL and SURFACE. Two important factors which are held to affect ASA estimates are 1) the different algorithms used by the programs, and 2) the choice of modified van der Waals radii used in the calculations (see Connolly (1996)).
From these examples it seems that both algorithms are capable of giving good estimates of the ASA, and give good agreement with each other. It also appears that better estimates are obtained using higher values for the ``precision parameters'' ZSTEP (in SURFACE) and PNTDEN (in AREAIMOL), as would be expected. It also seems that different choices of van der Waals' radii have a small effect on the ASA estimates, of a similar order of magnitude to the effect of varying the precision parameters.
The test cases chosen are far from comprehensive. Chothia examined a much larger number of cases (15 proteins) and remarked that ``the values of residue accessible surface areas [using the Lee and Richards algorithm] were similar to those found by Shrake and Rupley, though they used slightly different van der Waals' radii and averaged over a number of different residue confirmations.'' The results obtained here seem consistent with this conclusion.
Finally, the aim of this article was not to ``prove'' than one method or program is ``better'' than the other. Unsurprisingly, both programs seem to perform best when using the highest precision settings on ZSTEP/PNTDEN, and there seems little to choose between the two algorithms. Ultimately the choice of program will be determined by the type of problem being examined, based on the list of program functionalities in section 3.3.
AREAIMOL is still undergoing development, and at present the plan is to include the desirable features from SURFACE (which is no longer under active development) which are currently missing - for example, atom selection options and more powerful Van der Waals assignments.
Beyond that the possibilities are to extend the range of analyses offered by the program. One example is the inclusion (as of CCP4 4.0) of the facility to search for ``isolated surfaces'', which identifies surfaces which are completely enclosed inside the molecule (i.e. closed cavities at least as big as the probe sphere). Such cavities will contribute to the total accessible area of the molecule using the algorithms described in section 3.1, but in practice should not be counted as part of the external accessible surface. They may also be of interest in their own right, as features of the protein structure. Other possibilities under consideration include more comprehensive analyses of the location of area differences and buried atoms, or trying to identify features such as tunnels.
As always comments and feedback are welcome. If you have any suggestions for improvments or future developments then please address them to me at p.j.briggs@dl.ac.uk.
Connolly, M.L. (1996) "Molecular Surfaces: A Review", http://www.netsci.org/Science/Compchem/feature14.html
Chothia, C. (1975) Nature 254 304-308
Lee, B. and F.M. Richards (1971) J. Mol. Biol. 55 379-400
Richards, F.M. (1977) Annu. Rev. Biophys. Bioeng. 6 151-176
Sevcik, J., Dauter, Z., Lamzin, V.S. and Wilson, K.S. (1996) Acta Cryst. D 52 327-344
Shrake, A. and Rupley, J.A. (1973) J. Mol. Biol. 79 351-371