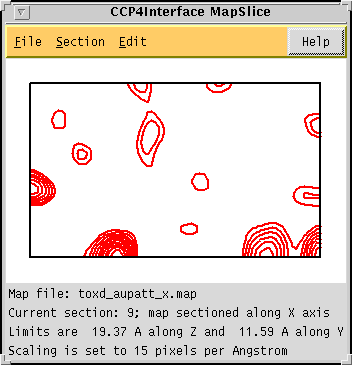
| The "mapslice" utility is still under development
and at present still lacks much of even the basic functionality required.
Ultimately the aim is to include the following:
|
The first official release of the CCP4 graphical user interface, CCP4I 1.0, was announced in April this year. Since then something like 180 sites have downloaded the software from the York FTP site. It's hard to know how many of the 180 sites are using the Interface for real - I've received feedback from 20-30 sites - some of it even complimentary! So are all the others using it happily without problems or have they not really got into it yet? Please let me know!
Peter Briggs and I have visited several crystallography labs in
the UK to do demos and run mini-workshops to help people get started using
the Interface and we would be very happy to do more so let us know if you
would like us to visit you for a day. We've also done demos
at conferences: the ACA in Buffalo, IUCr in Glasgow and the European Crystallography
Workshop in Como.
The interface has been very useful for workshops where participants
may be unfamiliar with CCP4 programs - we used it for the Refinement Workshop
in York in December 1998 (prior to full release of the software) and at
a recent International School in Barcelona.
Pete and I are now working on the enhancements to the Interface. Projects we are currently working on:
Interfaces to programs that got missed first time around - Sfall, Overlapmap, Detwin, Shelx and interfaces to some of the new programs entering the suite.
Enhancements to the Loggraph program particularly allowing the user to edit the graphs before printing them out for presentations or publications.
A Mapviewer which should replace NPO and xplot84driver for visualising map sections.
|
| The "mapslice" utility is still under development
and at present still lacks much of even the basic functionality required.
Ultimately the aim is to include the following:
|
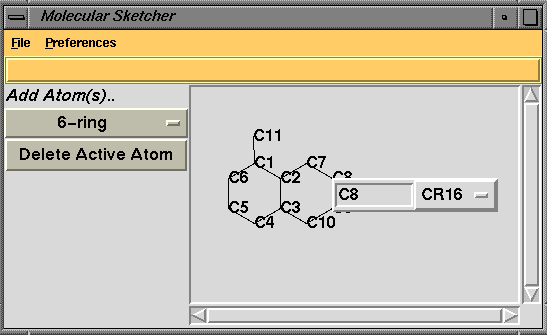
A Molecular Sketcher to simplify defining novel ligand molecules which need new entries in the refinement dictionary. This will go with the improvements to handling geometric constraints currently being worked on by Garib Mushudov and Alexei Vagin. An exampe of the Sketch window is shown below - the user has 'sketched' the structure and has clicked on the atom C8 to open a small window which enables editing of atom type and atom name. When the user has defined the molecule they will be able to automatically add the structure to their dictionary.
Porting the Interface to Windows NT. The basic Interface is now working on NT thanks largely to the Tcl/Tk language porting painlessly. There are a couple of more serious issues.
One problem I find working with the computer network setup at York is that the same file has different path names seen from Unix or NT machines (and you can get the same problem with an all-Unix system). I expect this is going to be a general problem as people will work in a 'mixed' environment. The interface already uses the idea of project directories and directory aliases and these need to be made a bit more flexible to cope with different machine domains.
The other possible issue with NT is that it does not support the rsh
command (or anything similar) which the Interface uses to run remote jobs
on Unix systems. The solution to this may be to run a 'CCP4I
server' on the 'remote' machine. Or will NT users expect to
always run jobs on their own machine? I would be interested to hear
from anyone who has any ideas on what would be useful or possible solutions.
See the Installation
Guide if you want to download the interface.