
By George M. Sheldrick
Institut für Anorganische Chemie, D37077 Göttingen, Germany
Fig. 1.
E-Fourier recycling as used in SHELXS-86 to improve phases from
direct methods.
Usually a couple of cycles were sufficient. Since the E-Fourier recycling was only applied to the 'best' solution, and only E-values greater than (say) 1.2 were employed, the computing requirements were modest. Very often this procedure was able to find every atom (except perhaps disordered solvent molecules), which users found very convenient. On a few occasions the E-Fourier recycling succeeded in extracting the solution from a rather dubious set of direct methods phases, but despite this strong hint, it did not occur to me that it could itself be effective as a 'direct method'. This required the development of the Shake & Bake philosophy by Weeks, Miller & Hauptman at Buffalo (Miller, DeTitta, Jones, Langs, Weeks & Hauptman, 1993; Miller, Gallo, Khalak & Weeks, 1994), which inspired much of the work reported here.
CC = [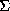 wEo2Ec2*w - wEo2*wEc2
] /
wEo2Ec2*w - wEo2*wEc2
] /
{ [wEo4*w - (wEo2)2] *
[wEc4*w - (wEc2)2 ]
}1/2
The correlation coefficient is more sensitive in the important early stages, and appears to give a very good indication of the true phase error (e.g. Fig. 2).
 <
<Tests on rubredoxin by Sheldrick & Gould (1995) showed that the elimination of atoms to improve the correlation coefficient (peaklist optimisation) was very effective at expanding the structure from the iron and four sulfur atoms to all ca. 400 atoms, provided that the resolution was better than 1.3Å.

Not yet available
Fig. 4.
(a) Sim-weighted E-map for a
cytochrome c6 (Frazão et. al, 1995)
with phases from the iron and three sulfur atoms;
the E-weighted mean phase error is 57deg.; (b) after peaklist
optimisation (E-weighted mean phase error 38deg.); (c) the final 3Fo-2Fc map after refinement of the structure.
Iterative application of peaklist optimisation enables about 90% of the protein atoms to be identified from the peaklist alone without the need to examine any maps; this was however required to find the remaining atoms, which had high thermal displacement parameters or were disordered.
Ec2(Eo2-1) for the largest E-values) for a known small
fragment (a small piece of  -helix proved very effective) or (b) threefold
Patterson superposition from vector triangles identified in the sharpened
Patterson peaklist (to exploit the presence of heavier atoms such as sulfur or
phosphorus). Since these two methods of generating slightly better than random
starting phases are not able to position the origin of the space group, all
calculations were performed on data expanded to the effective space group P1.
Expansion to P1 may in any case increase the chances of this approach
converging to the correct solution, but increases the computer time required.
-helix proved very effective) or (b) threefold
Patterson superposition from vector triangles identified in the sharpened
Patterson peaklist (to exploit the presence of heavier atoms such as sulfur or
phosphorus). Since these two methods of generating slightly better than random
starting phases are not able to position the origin of the space group, all
calculations were performed on data expanded to the effective space group P1.
Expansion to P1 may in any case increase the chances of this approach
converging to the correct solution, but increases the computer time required.

Tests showed that the peaklist optimisation was much more effective
than simply accepting the top N peaks, but that it takes about the same CPU
time as three structure factor calculations, and so is slower. Starting with
slightly better than random phases from the rotation search or Patterson
superposition map considerably increased the success rate of this approach.
The method was successful in solving several structures with more than 200
atoms in the asymmetric unit, but proved very expensive in consumption of
computer resources. The computer time required could be reduced considerably
by calculating the correlation coefficient for only the largest E-values, for
which structure factors were required anyway to provide initial phases for the
tangent refinement. However the correlation coefficient proved much less
effective when not applied to the full range of E-values. The solution was to
divide the procedure into an internal loop, in which a specified number
of peaks were eliminated so that Ec2(Eo2-1) remained as
large as possible, alternating with tangent phase refinement, and an
external loop, applied only for solutions with good values of CC (for
all data), in which peaklist optimisation as described above was applied
using all data so that the final structure was as complete as possible. The
new procedure (which has somehow acquired the name half-baked ) is
illustrated in Fig. 6.

In general, it appears to be computationally more efficient to expand the data to an effective space group of P1 for monoclinic structures; a larger percentage of trials lead to a solution, more than compensating for the increased cycle time. For higher symmetry it may be better to impose the full space group symmetry. It should also be possible to include twinning in the external loop; sometimes it is easier to guess the twin law than the space group, in which case the data could be expanded to P1.
The procedure described above is philosophically similar to Shake & Bake, but relative to Shake & Bake it does more of the work in real than in reciprocal space. It appears to be roughly comparable in its ability to solve difficult structures. One structure solved at about the same time by both programs, but using two different synchrotron data-sets, is vancomycin, a glycopeptide antibiotic of crucial medical importance in the struggle against the evolution of antibiotic resistant bacteria. The unexpurgated solution obtained by Schäfer, Schneider & Sheldrick (1996) using the half-baked procedure is shown in Fig. 7. The data were 99.3% complete to 1.09Å, the edge of the image plate used for synchrotron data collection. Including solvent there are 313 atoms in the asymmetric unit in P43212. 2000 trials with 8 cycles in the internal loop gave one solution; the CC of 75.5% was well separated from the rest (the next largest CC was 57.9%, for an incorrect solution). The CPU time used corresponded to a mere 4 VAX-years.

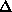 F or MAD FA values.
Patterson methods work well if there are only a few such atoms, but the
complexity increases as the square of the number of atoms and becomes
prohibitive, even for automated computer interpretation (Sheldrick et
al., 1993) when the number of anomalous scatterers is more than about 12.
One would have expected that classical direct methods should be able to solve
this problem, since they are capable of finding at least 100 equal atoms, and
the anomalous scatterers are usually separated from one another by distances
much greater than the limiting resolution of the reflection data, but in
practice they invariably fail to locate say 20 independent selenium atoms.
There seem to be several possible reasons for this unexpected problem.
F or MAD FA values.
Patterson methods work well if there are only a few such atoms, but the
complexity increases as the square of the number of atoms and becomes
prohibitive, even for automated computer interpretation (Sheldrick et
al., 1993) when the number of anomalous scatterers is more than about 12.
One would have expected that classical direct methods should be able to solve
this problem, since they are capable of finding at least 100 equal atoms, and
the anomalous scatterers are usually separated from one another by distances
much greater than the limiting resolution of the reflection data, but in
practice they invariably fail to locate say 20 independent selenium atoms.
There seem to be several possible reasons for this unexpected problem.
(a) Both Patterson and direct methods work best with complete data. Missing centric and other reflections cause problems.
(b) The F values represent lower limits on FH (MAD FA values should be better,
at least in theory), so small F values cannot be used in probability formulae
such as those involving negative quartets.
(c) It is difficult to take  (F) or (FA) into account in conventional direct and
Patterson methods, so the signal may get lost in the noise.
(F) or (FA) into account in conventional direct and
Patterson methods, so the signal may get lost in the noise.
(d) The selenomethionines may be conformationally disordered.
Table 1. Crossword table for the second best solution from the Cu-K
anomalous F values for a HiPIP protein with two Fe4S4 clusters in the
asymmetric unit (1.5Å data kindly donated by Hazel Holden & Gary
Wesenberg, truncated to 2Å to make the test more difficult). The upper
row gives the minimum distance between the atom defining the row and the atom
defining the column, the lower row gives the corresponding Patterson
superposition minimum function.
Try 89, CC(HA)=35.74%, PATFOM=39.67
Peak x y z self cross-vectors
99.9 0.389 0.736 0.176 29.2
41.0
98.4 0.432 0.746 0.249 30.1 2.6
51.0 66.6
90.7 0.399 0.696 0.194 29.4 2.2 3.3
0.0 47.5 33.0
89.9 0.914 0.187 0.126 27.9 14.0 16.6 14.4
53.2 34.6 49.1 74.7
88.1 0.354 0.742 0.255 31.4 2.6 2.9 3.4 14.6
45.7 69.3 73.2 56.5 57.4
82.3 0.960 0.160 0.043 26.6 14.6 17.0 14.8 3.2 14.7
67.6 42.5 37.9 54.7 27.5 37.8
71.1 0.901 0.125 0.082 27.7 14.0 16.5 13.8 3.5 14.5 3.0
22.2 27.9 32.6 34.9 25.3 32.8 47.8
67.4 0.973 0.342 0.132 27.4 16.6 18.8 18.0 8.4 16.8 9.9 11.8
41.7 0.9 49.0 20.0 0.5 31.5 0.0 0.0
46.8 0.966 0.143 0.145 27.6 16.4 18.9 16.5 3.1 16.8 3.0 3.1 10.4
38.3 34.2 43.8 19.7 22.5 25.3 26.7 45.8 0.0
41.3 0.500 0.749 0.286 28.8 5.1 2.7 5.3 19.1 5.4 19.6 18.9 21.3
0.0 4.5 46.5 0.7 14.7 21.6 5.8 2.8 22.7
Direct methods based on real/reciprocal space recycling have some advantages
to offer that may help to overcome these problems. The number of anomalous
scatterers NH is usually known precisely; this information can be used in a
very direct way. The elimination of atoms in turn to optimise the correlation
coefficient CC, until exactly NH atoms remain, does not require complete data.
In addition CC incorporates weights based on the experimental sigmas. Finally,
the Patterson function can still be used as an independent check, as shown in
Table 1. The second best solution is illustrated; the Patterson superposition
minimum function values clearly show that the atoms 1-7 and 9 correspond to the
eight expected iron atoms. They form two Fe4 clusters with Fe***Fe distances
of about 3Å. The PATFOM figure of merit is simply the mean of the
Patterson superposition minimum function values for the top NH atoms. The
solution with the best PATFOM, but the second best CC, gave atoms 1-8 as the
correct iron atoms.
Table 2. Crambin test, internal loop searching for 3 disulfide bonds, external loop expanding to full structure. The 0.92Å low-temperature data were collected and provided by Håkon Hope.
1625 E-sig(E) > 1.500 used to generate 77607 unique TPR
Try 19, CC(HA) = 19.03%, PATFOM = 13.80
Peak x y z self cross-vectors
99.9 0.3019 0.1253 0.1020 19.2
15.6
96.7 0.2571 0.0783 0.1028 22.4 2.0
18.4 14.3
96.7 0.3914 0.1707 0.4511 13.0 8.6 9.6
18.2 13.8 18.4
93.7 0.4373 0.1292 0.4262 11.1 9.1 10.3 2.1
16.3 35.5 12.2 11.3
90.7 0.0794 0.2353 0.0483 11.5 9.4 7.9 15.5 16.9
0.9 17.0 15.6 10.4 12.9
85.7 0.1098 0.3147 0.0591 13.1 8.6 7.5 14.6 16.0 1.9
13.2 11.5 10.3 16.1 7.7 0.0
Peaklist optimization cycle 1 CC=30.05% for 41 atoms
Peaks: 99 97 97 93 92 88 15 15 -14 14 -13 -12 -12 -12 -12
Peaklist optimization cycle 2 CC=47.75% for 108 atoms
Peaks: 99 95 93 92 85 81 34 -34 34 34 34 -33 33 33 33 33
Peaklist optimization cycle 3 CC=70.62% for 240 atoms
Peaks: 99 95 91 90 80 77 36 36 36 35 35 35 34 34 34 33 33
Peaklist optimization cycle 4 CC=81.57% for 354 atoms
Peaks: 99 96 92 88 74 73 37 37 37 36 36 35 35 35 35 35 34
Fragments: 310 2 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1
It is possible to combine the search for a specific number of heavier atoms from the native data in the internal loop with expansion to the full structure in the outer loop, as illustrated by the crambin test in Table 2. In this case the three disulfide bridges can be identified by their distances of about 2Å; only solutions containing three disulfide bonds were expanded further by the program. The external loop of peaklist optimisation leads to the essentially full structure in 4 cycles with a convincing CC (values greater than 70% are invariably correct). A minus sign in the list of peak heights indicates that that peak was rejected in the elimination procedure. The final line shows that there is a connected fragment of 310 atoms, plus a number of well-defined water molecules that do not bond to other atoms.
To extend the method to lower resolution, density modification may not prove sufficiently incisive as a replacement for peak-picking. More promising is the method used in ARP (Lamzin & Wilson, 1993) to fill density with atoms. Alternatively, instead of using individual atoms, typical groups of 3-5 atoms (e.g. peptide units) could be fitted to the density (given a fast computer !).
It looks as though real/reciprocal space recycling has the potential to overcome the current difficulties in the location of a large number of anomalous scatterers from noisy MAD data, but this needs further testing on real data.
I am grateful to the Fonds der Chemischen Industrie for support. Figs. 1, 2 and 5 are reproduced from Sheldrick & Gould (1995), Fig. 4 from Frazão et al. (1995) and Fig. 7 from Schäfer et al. (1996), with permission of the respective publishers.
References
Frazão, C., Soares, C.M., Carrondo, M.A., Pohl, E., Dauter, Z., Wilson, K.S., Hervás, M., Navarro, J.A., De la Rosa, M.A. & Sheldrick, G.M. (1995). Structure 3, 1159-1169.
Fujinaga, M. & Read, R.J. (1987). J. Appl. Cryst. 20, 517-521.
Lamzin, V.S. & Wilson. K.S. (1993). Acta Cryst. D49, 129-147.
Miller, R., DeTitta, G.T., Jones, R., Langs, D.A, Weeks, C.M. & Hauptman, H.A. (1993). Science 259, 1430-1433.
Miller, R., Gallo, S.M., Khalak, H.G. & Weeks, C.M. (1994). J. Appl. Cryst. 27, 613-621.
Read, R.J. (1986). Acta Cryst. A42, 140-149.
Schäfer, M., Schneider, T.R. & Sheldrick, G.M. (1996). Structure 4, 1509-1515.
Sheldrick, G.M. (1982). In Crystallographic Computing, edited by D. Sayre, pp. 506-514. Oxford: Clarendon Press.
Sheldrick, G.M. (1990). Acta Cryst. A46, 467-473.
Sheldrick, G.M., Dauter, Z., Wilson, K.S., Hope, H. & Sieker, L.C. (1993). Acta Cryst., D49, 18-23.
Sheldrick, G.M. & Gould, R.O. (1995). Acta Cryst. B51, 423-431.