See also the accompanying document giving background information.
This is an introduction to the procedures for using Refmac5 to refine a crystal structure.
See also the documentation for Refmac5 and Sketcher.
In the following instructions, when you need to type something, or click on something, it will be shown in red. Output from the programs or text from the interface is given in green.
This example is to refine the protein RNAse Sa in its unliganded and liganded form, for which we know:
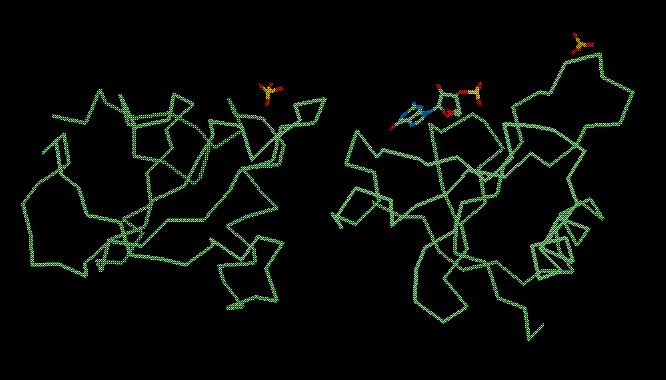
The final structure (solved by Joseph Sevcik: J. Sevcik, Z. Dauter, V.S. Lamzin, K. Wilson, Acta Cryst. D52 (1996) p327-344) looks like this:
There are 1749 atoms in the asymmetric unit. If we describe each atom using three positional parameters x,y,z and an isotropic temperature factor B there are 6996 parameters. In the experimental data (see $DATA/rnase18.mtz) there are 17991 reflections giving an observation-to-parameter-ratio of 17991/6996 = 2.57. This is not enough to refine all parameters as independent variables. However we have a great deal of information about the geometry of molecules - the bond lengths and bond angles etc. The refinement program will set up restraints between related atoms which say, for example, that the distance between two bonded atoms must be close to the ideal bond length.
Refinement programs read libraries describing the expected geometry. These contain information about the ideal bond lengths, bond angles, planar groups etc. for the common chemical monomers which are found in macromolecules (e.g. amino acids and nucleic acids). If this information is incorrect you will not get a correct structure.
If your structure contains an unusual substrate molecule or a modified amino acid, then there may be no suitable description in the library, and you will have to provide one in the required style. This can be difficult; it is essential you know the chemical definition of your ligand, (e.g. which atoms lie in a plane, which bond type exists, etc). This knowledge must then be written in the correct format for the refinement program to read. This can also be challenging, so we will make the geometry description for the ligand to show how it is done.
Files in directory DATA:
| rnase.pdb | The refined unliganded protein coordinates |
| GMP.pdb | Idealised coordinates of the ligand molecule, guanosine-3*-monophosphate |
| rnase-3gp.pdb | The refined protein-ligand coordinates |
| rnase_bad.pdb | A test set of coordinates with errors introduced. |
| rnase18.mtz | An MTZ file containing three sets of experimental data extending to 1.8Å, labelled FNAT SIGFNAT F3GP SIGF3GP F2GP SIGF2GP for unliganded protein, the protein liganded with guanosine-3*-monophosphate (3GP) and with guanosine-2*-monophosphate (2GP), respectively | rnase115.mtz | The experimental data and calculated structure factors and phases for the unliganded protein to 1.15Å |
| rnase25.mtz | An MTZ file containing experimental data (including anomalous), extending to 2.5Å, for the native protein and three derivatives (mercury, platinum and iodine), as used for experimental phasing by MIR and experimental phasing by MAD |
The output files in directory RESULTS:
| 3GP_mon_lib.cif | The monomer library description of 3GP |
| review.log | The log file from running Refmac5 to review restraints |
| rnase18_bad_refmac1.pdb | The output file from running Refmac5 to review restraints |
| refmac-unliganded.log | .log from Refmac5 refinement of unliganded RNAse |
For the tutorial a file called $DATA/rnase_bad.pdb bas been generated, which has had some errors introduced. The residue ARG B:63 is now too close to ARG A:63. Also, all residues have been shifted by up to 0.2Å.
We will use REFMAC5 in review restraints mode. The program will look at the atom coordinates and decide where there are disulphide bonds, cis-peptides and D-peptides. It will also calculate the distance between atoms and if they are very close it will assume the atoms are bonded and will make a make a restraint to say 'these two atoms must stay close'. Of course this is not always right. It will also add any absent atoms - if a residue does not have the right atoms it will make them. REFMAC5 will help you by finding the disulphide bonds etc. automatically but you MUST check that they are correct.
From the Refinement module select Run Refmac5.
In the Protocol folder, enter a Job title such as:
Then
Now select the input coordinate file:
Look in the folder called Setup Restraints. In here you can decide what to look for in the PDB file. We will use the defaults - you do not need to change anything.
Now run the REFMAC5 program. From the Run menu at the bottom of the window choose Run Now.
The job will take a little time. When it has finished, the job status will be "FAILED" - this is caused by WARNINGs (see below) which should be considered serious, but which are part of the learning process here. Look at the log file (click on the name of the job, refmac5, in the main window and use View Files from Job and View Log Files).
Some interesting things in the log file:
WARNING : CIS peptide bond is found, angle = 16.01
ch:AA res: 26 GLY --> 27 PRO
....
WARNING : link:SS is found dist = 2.211 ideal_dist= 2.031
ch:BB res: 7 CYS at:SG .->BB res: 96 CYS at:SG
These things are correct, REFMAC5 has checked the input protein molecule and found some cis peptide bonds and some disulphide bonds, but there is also:
WARNING : description of link:ARG-ARG not found in the dictionary.
link will be created with bond_lenth = 1.400
This is not true, REFMAC5 will suggest making bonds between the residues which are too close.
Now look at the output PDB file: use View Files from Job and select rnase_bad_refmac1.pdb (this file can also be found in the RESULTS directory).
At the top of this file is new information:
LINK NH1_ ARG A 40 NE__ ARG B 63 ARG-ARG LINK NH1_ ARG A 40 CZ__ ARG B 63 ARG-ARG1 LINK NH1_ ARG A 40 NH2_ ARG B 63 ARG-ARG2 LINK GLY A 26 PRO A 27 PNCIS SSBOND 1 CYS A 96 CYS A 7 LINK GLY B 26 PRO B 27 PNCIS SSBOND 2 CYS B 96 CYS B 7
Now Quit from the window.
It is necessary to edit the PDB file to remove the bad link information. There is an easy way to do this. From the Refinement menu in CCP4i main window select Edit Restraints in PDB.
Select the input file:
Wait while the program reads the file.
In the window you will now see:
The space group and the symmetry operators for the space group (you may need this information to define disulphide bonds or links between molecules that are not in the same asymmetric unit).
The MODRES IDs and LINK modes provide additional information to describe the molecule geometry. Definitions of MODRES allow you to modify a standard residue description, e.g. to rename a monomer, or to modify MET to include Se - the details are discussed in the Refmac5 documentation. LINK definitions describe ways to link two monomers, e.g. peptides co-valently linked to substrates.
This is a way to redefine non-standard residues. There is a monomer labelled GMP in the RNAse coordinate file, which matches the dictionary definition of 3GP.
The two disulphide bonds in RNAse are shown.
The three bad bonds are listed.
The two cis-peptides in RNAse are listed.
You can delete the bad links by clicking on the menu Edit Table and selecting Delete Last Row. Do this three times.
It is also possible to add new things - try clicking on Add Row.
We will stop using the rnase_bad coordinates, so you do not need to save changes - Close the window.
From the Refinement module select Run Refmac5.
Enter a suitable job title such as
Then
Also you will see:
If you have a graphics program to look at the maps then click this on and select a map format.
Now select the input files - the experimental data:
and make sure you have correct data columns:
and the coordinate file:
Click on Run -> Run Now.
The job will take a little time. When it is finished look at the log file (click on the name of the job, refmac5, in the main window and use View Files from Job and View Log Graphs).
If you do not have a log file then click on View Any File and set:
and then select file:
Go to the last table in the Tables in File and click on:
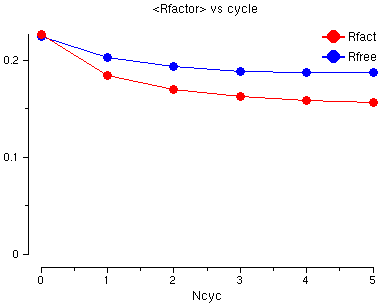
You will see a graph of the R factor and the Free R factor for the 6 cycles of refinement. The R factor is very good already but both go down a little.
Also look at the Graphs in Selected Table for:
The FOM tells you how well the molecule matches the experimental data and the Geometry tells you how well the molecule obeys the geometry restraints.
Also, slightly up the Tables in File list, select the last:
This is information about the last cycle of refinement. Have a look at:
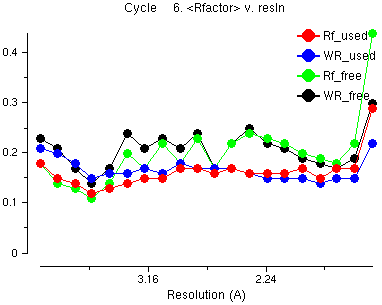
The red line is the average R factor versus resolution for the data which is used and the green line is the Free R factor (for the 'free' data which is not used). This is similar across the resolution ranges - it does not go up for high resolution data. This is an example of what is good about maximum likelihood refinement compared with the old-fashioned least squares.
Also look at the graph:
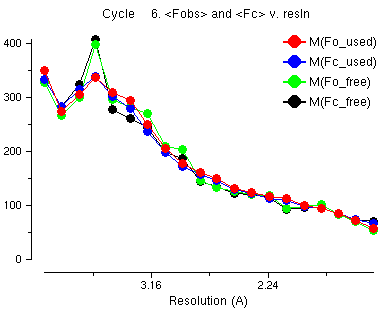
This is a graph of the average observed structure factors and calculated structure factors. You notice that at low resolution the observed (red) and the calculated (blue) are not the same. At low resolution the water atoms, which we can not see in the crystal structure, are an important part of the structure factors. The refinement program tries to model the water atoms by solvent scaling but it is difficult for this data because some of the very low resolution data is missing.
To close the loggraph window click on the File menu and select Exit.
Look at the header of the output MTZ file - click on View Files from Job and select the file rnase18_refmac1.mtz. In the file you will see:
* Column Labels : H K L FNAT SIGFNAT FreeR_flag FC PHIC FWT PHWT DELFWT PHDELWT FOM
The new data in the file is:
| FC & PHIC | the structure factors and phases calculated from the final coordinates |
| FWT & PHWT | the 'best' structure factors and phases weighted by the maximum likelihood function |
| DELFWT & PHDELWT | the 'best' structure factors and phases for a difference map |
| FOM | figure of merit for PHIC |
If you selected the option to create output maps then you can look at the maps created from the REFMAC output.
| ...FWT.map | the 'best' weighted map |
| ...DELFWT.map | the 'best' weighted difference map |
An example of these maps is shown below for a tyrosine residue which is in the wrong place. The DELFWT map is the weighted difference map of F(observed) - F(calculated) and looks like this:
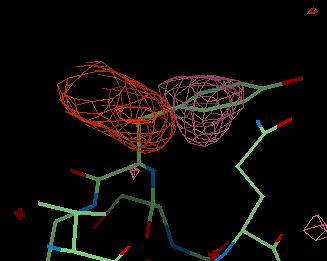
Here you can see a large pink area of negative density where the tyrosine side chain is now. This is saying that the side chain should not be here. The large brown-red area of positive density is showing where the side chain should be.
The FWT map is the weighted map and looks like this:
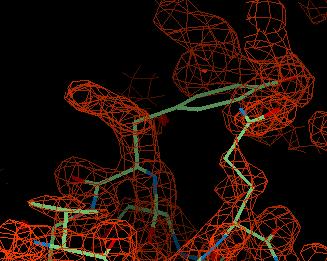
You can see region of density to the left of the tyrosine which is where it should go.
There are three ways to get a geometry description:
From the Refinement module select the Monomer Library Sketcher task.
From the File menu at the top of the window select Read File and from the next menu select Load Monomer from Library.
In the Load Monomer from Library window, in the Choose Monomer folder, search:
and select 3GP GUANOSINE-3*-MONOPHOSPHATE.
Select Run -> Run Now, and Close.
You will see the molecule displayed. You can rotate it by holding down the left mouse button. On the right of the window is a list of atoms - this list has the element, the atom name and the oxidation state (the charge of the atom). Below the list of atoms is a list of the chiral centres found in the molecule, of which there are four.
Now look at the monomer library file. In the Main Window select the last job which is called load_monomer. Now select the View Files from Job menu (on the right side of the main window) and select the file 3GP_mon_lib.cif.
In this file you will see a list of the atoms.
_chem_comp_atom.comp_id _chem_comp_atom.atom_id _chem_comp_atom.type_symbol _chem_comp_atom.type_energy _chem_comp_atom.partial_charge _chem_comp_atom.x _chem_comp_atom.y _chem_comp_atom.z 3GP O6 O O 0.000 0.000 0.000 0.000 3GP C6 C CR6 0.000 0.831 0.906 0.061 3GP C5 C CR56 0.000 2.177 0.611 0.120 ......
Further down the file is the list of bonds:
_chem_comp_bond.comp_id _chem_comp_bond.atom_id_1 _chem_comp_bond.atom_id_2 _chem_comp_bond.type _chem_comp_bond.value_dist _chem_comp_bond.value_dist_esd 3GP C6 O6 aromatic 1.230 0.020 3GP C6 N1 aromatic 1.380 0.020 3GP C5 C6 aromatic 1.390 0.020 3GP C4 C5 aromatic 1.390 0.020 ......
The words at the top of the list tell you what is in each column:
There is similar information on bond angle, torsion angle, chirality and planar groups.
The refinement program will try to make the ligand as defined in this file - you can edit the file if you need to.
From the Refinement module select the Monomer Library Sketcher task.
For the next steps see the view_sketcher_1 picture.
From the File menu at the top of the window select Read File and from the next menu select Read PDB file. Select the file:
If there is a message querying the use of an existing library file (using LIBCHECK), click No.
You will see the molecule displayed. You can rotate it by holding down the left mouse button. On the right of the window is a list of atoms - this list has the element, the atom name and the oxidation state (the charge of the atom). Below the list of atoms is a list of the chiral centres found in the molecule, of which there are four.
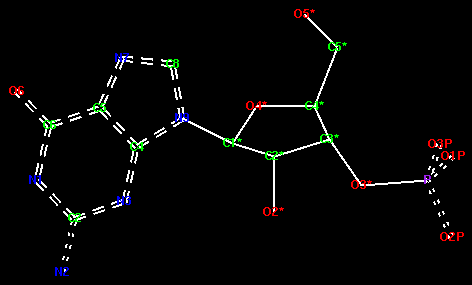
The picture of GMP below shows the correct delocalised and aromatic bonds - edit your molecule accordingly. To change a bond to a delocalised bond, you must hold down the Shift key on the keyboard and click on the bond with the right mouse button. It will step through single--double--triple--deloc--aromatic--metal.
Now create the monomer library. From the File pull-down menu at the top of the window select Create Library Description.
In the Create Dictionary Entry window, enter a job title such as
Select
Then enter the name of the ligand (this must be GMP which is the name of the ligand in the PDB file which we will refine). So:
The names of files will be created automatically so you can then select Run -> Run Now. Then Close this window.
You must wait a little time - a program called LIBCHECK is running. When it has finished the molecule is displayed again.
Now look at the new monomer library file. From the Main Window select the last job which is called dictionary. Select the View Files from Job menu (on the right side of the main window) and select the file GMP_mon_lib.cif.
In this file you will see a list of the atoms.
_chem_comp_atom.comp_id _chem_comp_atom.atom_id _chem_comp_atom.type_symbol _chem_comp_atom.type_energy _chem_comp_atom.partial_charge _chem_comp_atom.x _chem_comp_atom.y _chem_comp_atom.z GMP O31 O OP -0.660 0.000 0.000 0.000 GMP P3 P P 0.000 1.175 0.940 0.095 GMP O33 O OP -0.660 1.871 0.745 1.419 ......
Further down the file is the list of bonds:
_chem_comp_bond.comp_id _chem_comp_bond.atom_id_1 _chem_comp_bond.atom_id_2 _chem_comp_bond.type _chem_comp_bond.value_dist _chem_comp_bond.value_dist_esd GMP P3 O31 deloc 1.510 0.020 GMP O32 P3 deloc 1.510 0.020 GMP O33 P3 deloc 1.510 0.020 GMP O3* P3 single 1.610 0.020 ......
This list is not quite the same as for the monomer 3GP as it is in the Monomer Library. Have a look at the differences and update as you see fit.
This section is optional. Alternatively you can go directly to the next step - Review Special Restraints for ligand.
If you have no coordinates or other definition of the ligand then you must draw the molecule in the Sketcher. Sometimes there may be a similar molecule in the library - you can start from this and edit it. There is a guanosine molecule in the library which we can use to make GMP (or, more accurately, 3GP).
Delete any molecule that you have displayed: from the Edit pull down menu select Delete All Atoms.
From the File pull-down menu select Read File and then Load Monomer from Library. In the new window, from the folder Choose Monomer, select:
Now you will see a list of RNA monomers. Click on the line:
Then click on Run -> Run Now and Close the Load Monomer from Library window.
You must wait a little while before the molecule of guanosine is displayed. To make 3GP you must delete the phosphate group on the O5* and draw a phosphate group on O3*.
For the next steps see the view_sketcher_2 picture.
Make sure that the Mouse Mode (on the left of the Sketcher window) is set to Edit Monomer.
From the edit tools on the left of the Sketcher window select the
'Delete atom' icon ![]() from the edit tools on the left of the window.
from the edit tools on the left of the window.
Hold down the Shift key and click with the left mouse mutton on the atoms O3T, O2P, O1P and P to delete them.
To add the new phosphate group select the the 'Add a C atom' icon
![]() from the edit tools. The atoms that you add will be
carbon atoms - you will change them later to phosphorus and oxygen.
from the edit tools. The atoms that you add will be
carbon atoms - you will change them later to phosphorus and oxygen.
Now look at the end of the table on the right side of the Sketcher window. The new atoms are C21, C22, C23 and C24, and each atom has the elment type C. Change C21 to a P and the other three to O. The atom names are also wrong - change the names to P, O1P, O2P and O3P.
The bonds within the phosphate are delocalised bonds, so click Shift - right mouse button on the bonds between P and all three O atoms, stepping through the bonds until they are 'deloc' bonds.
Now we create the monomer library. From the File pull-down menu at the top of the window select Create Library Description. In the window enter the name of the ligand (call this TEST3GP so you do not overwrite the files you made before). So:
The names of files will be created automatically so you can then click Run -> Run Now. Close this window.
Wait while the program runs to build the dictionary file. The molecule is drawn again. If necessary you can make corrections and run again. To close the Sketcher window, select the File pull-down menu and Close Sketcher.
We will add the ligand to our refined unliganded structure. In order to see what might happen, a file called $DATA/rnase-3gp.pdb is provided, with the refined unliganded model plus 3GP.
We will use REFMAC5 in review restraints mode. The program will look at the atom coordinates again. The disulphide bonds, cis-peptides and D-peptides are already defined. It will also calculate the distance between atoms and if they are very close it will assume the atoms are bonded and will make a make a restraint to say 'these two atoms must stay close'. Of course this is not always right.
From the Refinement module select Run Refmac5.
Enter a suitable job title such as
Then
Select the input coordinate file:
Select the new library file that you made for 3GP (this can also be found on the RESULTS directory):
Now run the REFMAC5 program. From the Run menu at the bottom of the window choose Run Now.
If all is well, the program should run without any warnings, apart from those about hydrogens. Now we are ready for some real refinement.
Now we will use the monomer library description that we created to refine the rnase molecule with the 3GP ligand.
From the Refinement module select Run Refmac5.
Enter a suitable job title such as
Then
Also you will see:
If you have a graphics program to look at the maps then click this on and select a map format.
Now select the input files - the experimental data:
and make sure you have correct data columns:
and the coordinate file:
To use the geometry description file which you have made:
Click on Run -> Run Now.
The job will take a while. When it is finished look at the graphs from the log file (click on the name of the job, refmac5, in the main window and use View Files from Job and View Log Graphs).
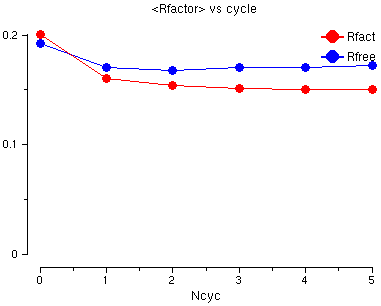
Go to the last table in the Tables in File and click on:
You will see a graph of the R factor and the Free R factor for the 6 cycles of refinement. The R factor is very good already but both go down a little.
To close the loggraph window click on the File menu and select Exit.
Back to the previous tutorial - Molecular Replacement.
Back to the index.
To find out more:
Refmac: http://www.ysbl.york.ac.uk/~garib/refmac
Libcheck: http://www.ysbl.york.ac.uk/~alexei/libcheck.html
CCP4: http://www.dl.ac.uk/CCP/CCP4
Prepared by Liz Potterton (lizp@ysbl.york.ac.uk) and Eleanor Dodson, July 2000
Adapted by Maria Turkenburg, 2002-2003