Gesamt aligns two or more structures using the algorithm of efficient clustering of short fragments, where the fragments are made from adjacent protein backbone C-alpha atoms, followed by an iterative three-dimensional refinement based on a dynamic programming procedure.
Gesamt uses the pairwise alignment algorithm when comparing two structures or looking for structural hits in PDB archive. When more than 2 structures are given on input, Gesamt uses the multiple alignment algorithm. Note that multiple alignment does not reduce to a set of pairwise alignments. Multiple alignmet is usefu for the identification of common structural motifs in whole protein families.
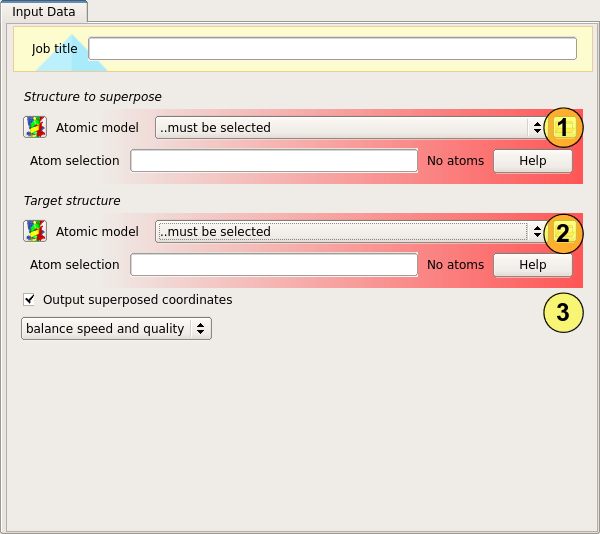
Input
The superposition requires two sets of atomic coordinates. The first set of coordinate (1) are the model to be fitted - this model will be rotated and translated to place it on top of the other model. The second set of coordinates (2) are the target model - this model will not be moved.
In each case a coordinate selection may optionally be specified to allow a subset of the model to be used.
The remaining options (3) control whether the moved model will be saved (otherwise only a report is generated), and how the matching will be performed. The default mode balances the quality of alignment and computation speed. This is a recommended mode for most applications. In high quality mode, Gesamt attempts to reach maximal quality with no reference to speed considerations. In this mode, Gesamt is about 10 times slower and achieves quality improvement in few percents of all instances on comparison with normal mode.
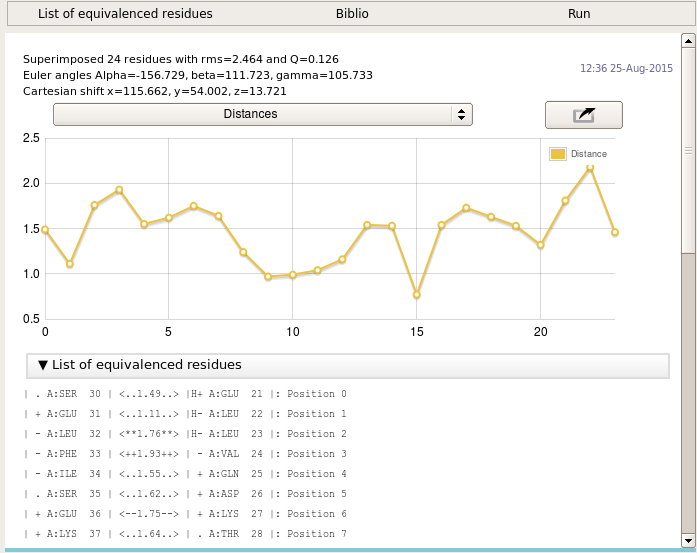
Gesamt reports the Transformation Matrix calculated for superimposing foo_1st.pdb onto foo_2nd.pdb, Q-score and the RMSD from the superposition, as well as polar and Euler rotation angles and orthogonal translation vector.
The distance graph shows the distance between the atoms of the aligned residues.
The program then gives a residue-by-residue listing of the alignment. Strands and helices in the two structures are identified and given in the output. The output also lists distances between all matched residues at best structure superposition.