Contents
IntroductionSecondary Structure (SSM)
Match Close Residues
User defined matches
Transforming Coordinates
Saving the transformed coordinates
Methodology
 |
CCP4 Molecular Graphics Documentation | |
| Superpose |
| Documentation Contents | On-line Documentation | Tutorials | CCP4mg Home |
Access to the Superpose application is from the Application menu. There are three different methods for superposing models which are chosen from the box at the top of the Superpose window:
SMM - automatic secondary structure matching to superpose the equivalent SSEs of proteins
Match Close Residues - will find equivalent atoms of a roughly superposed structure; this is useful to fine tune superposition of a region of structure such as an active site
User defined matches - the user must specify the matching atoms or residues - this method includes an algorithm to find a best match of similar ligands.
These methods are explained in the folllowing sections.
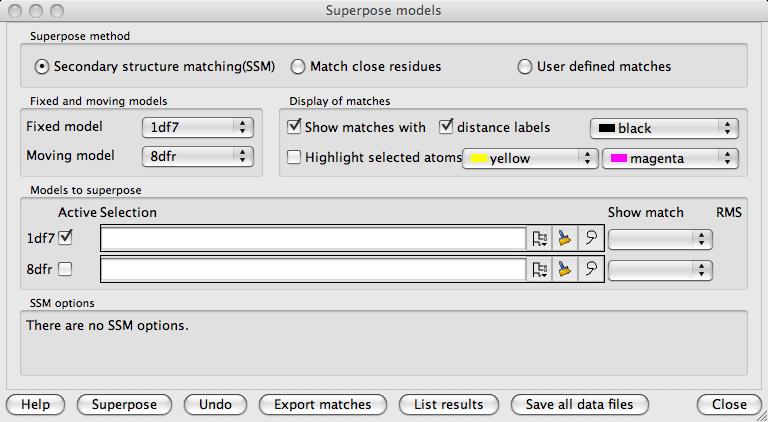
In the second box down in this window is a choice of the Fixed model and a highlighted Moving model. All other models will be superposed on the fixed model. All graphical display of matches, distances and selections are shown for the fixed model and the one highlighted moving model.
In the Display of matches box are options controlling what is displayed in the main graphics window. The displayed features are always for the fixed model and the highlighted moving model. If Show matches is checked the matched atoms are shown by dashed lines and optionally labelled with the inter-atomic distances. If Highlight selected atoms is checked and a limited set of atoms has been selected for superposition then these are indicated by coloured 'pyramids' on the selected atoms.
The Models to superpose box is visible if the SSM or the Match close residues options are selected. All of the loaded models are listed with a line for each model containing:
The Active checkbox controlling whether the model is to be superposed
A Selection widget (see selection widget) to select a limited set of atoms to be used in the superposition (the default is 'All' if there is no specified selection)
The Show match menu appears after SSM superposition and enables viewing alternative superpositions.
The RMS lists the RMS difference and the number of matched atoms after models have been superposed.
There is an Options box at the bottom of the window which changes appropriately for the selected superposition method.
The buttons at the bottom of the window:
Superpose superposes the active structures by the selected method.
Undo move the structures back to their original coordinates.
Export matches will write a list of the matched atoms (as shown is the Show matches checkbox is checked) to a vector file which can then be read in as a Vector object. This may be useful if your are creating a publication picture as Vectors can be edited and have more flexible display styles.
List results will give a listing of the matched SSEs (secondary structure elements)
and residues, the inter-atomic distances and the transformation matrix. In the listing window there is an option to save the information to a file.
Save all data files Will save all of the PDB files with the same tag added to the file name.
This method will superpose protein structures very quickly and without needing any input from the user to specify equivalent residues. The method works equally well for close homologs or for matching limited regions of similarity in very different structures. The Selection tool can be used to select limited regions of the loaded models for matching but note that the method works by first matching equivalent secondary structure elements so is not applicable to very small fragments of protein or non-protein which do not include a α-helix or β-strand. If the user's selected atoms include one Cα from a SSE then the whole of the SSE will be used in the superposition.
For some structures there may be several possible superpositions which score similarly so the Show match tool can be used to view other possible superpositions.
The methodology is described below.
This method is useful for performing locally optimised superposition after a global superposition by SSM. For example this method will optimise the superposition of the residues forming the binding site of a ligand. To do this you need to select the required local region of the 'fixed' model. This is probably best done using the Neighbourhood of.. option from the 'fixed' model's Selection tool and selecting the Neighbourhood of the ligand. It is not necessary to make any atom selection for the 'moving' models.
This method will attempt to find the equivalent residues in the 'moving' model to the selected residues in the 'fixed' model. The criteria for equivalent residues is that the Cα atoms are within a given cutoff distance and the residues have similar orientation. The equivalent atoms within the equivalent residues must have the same name and be within a given cutoff distance. A least squares fit of all equivalent atoms is performed.
For this method the user has several means of specifying equivalent atoms, residues or ranges of residues to superpose. Monomers (i.e. not amino acid or nucleic acid) can be matched and the equivalent atoms will be found even if the monomers are not identical.
For this superposition method the Superposition interface is significantly changed to show a User defined matches frame
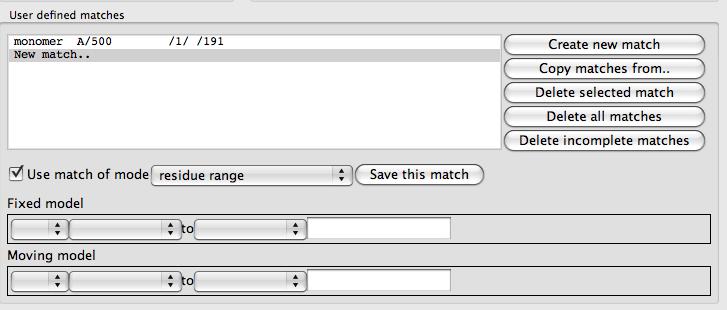
You must select two models to work on at any one time: a Fixed model and Moving model. Selecting the matching atoms or residues can be done via the User defined matches frame or via tools on the popup menu that appears if you click on an atom in the fixed or moving molecule with the right mouse button.
The User defined matches frame contains a list of the matches between the fixed and moving model and some push button commands to the right of the list. There is a tool to enter or edit matches below the list. The buttons are
Create new match will clear the match edit tool for you to enter a new match.
Copy matches from if you have already set up matches for a different pair of fixed and moving models then this tool will enable you to copy the same matches o use for the current pair of models. But note that in the User defined matches options further down the window the option to apply the superpose to all active models may also serve your purpose.
Delete selected match will delete the match curently seelcted in the list of matches
Delete selected match will delete all matches
Delete incomplete matches will delete any matches that have not been fully specified with selections for both fixed and moving model
The match editor has a checkbox labelled Use match which can be unchecked to switch off the match (but the selections will still be saved for future use). The match mode combo box allows selection of the match mode. The possible match modes are:
atom a single atom in the fixed model is matched to a single atom in the moving model
residue a single residue in the fixed model is matched to a single residue in the moving model. The option in the User defined matches options frame specifies which atoms in the residue will be matched (the default is CA atoms).
residue range a range of residues in the fixed model is matched to a range of residues in the moving model. The option in the User defined matches options frame specifies which atoms in the residues will be matched (the default is CA atoms).
monomer This match mode is most appropriate to matching residues which are neither amino acid nor nucleic acid. The matched monomers do NOT have to be identical. The method uses a graph matching algorithm to find the best equivalent atoms in the matched monomers.
selection This match mode uses a different style of widget in the match edit tool. The selection widget allows a more general selection but note that you must select the same number of atoms in the fixed and the moving model and that they will be matched according to their order in the PDB file.
close atoms in selection This match mode also uses the selection widget. This match mode will match only those atoms close enough to satisfy the criteria described for the Match close residues method using the parameters in the Close residues options frame (this frame is only visible when the Close residues method is selected). For this match mode it is necessary to have the models roughly superposed but it is acceptable to only enter a selection for one of models.
Below the match mode line in the match editor are two widgets to select the set of atoms in the fixed model and the moving model. After specifying the match remember to click the Save match button. Only the matches that appear in the list and are not greyed out (indicating they are inactive) are used when you click the Superpose button for a least-squares best fit of all matched atoms.
The alternative means of specifying matches uses the popup menu which appears when you click on an atom with the right mouse button. This menu has items labelled Superpose with the appropriate options from these:
Select this.. and a sub-menu of options atom,residue, start residue range etc.. After you have selected one of these, the required selection will be highlighted on the model with 'pyramids'. If you select 'Start of residue range' then you will need to click again on an atom to select 'End of residue range'. You can make several selections in either the fixed or the moving molecule before either selecting the matches in the other molecule (the Match to.. option below).
Match to.. and a sub-menu listing selections is available after one or more selections have been made for the other molecule. If one of the selections is chosen then the apropriate atoms are matched and the matches are indicated by dashed lines between the atoms. Note that for matching residue ranges the the option in the Superpose window For residue range match .. atoms is applied.
Undo last selection/match will undo the last command.
Saving matches The matches are saved to the ccp4mg file for both the fixed and moving model. This data will be retrieved by the Superpose application whenever both models are laoded.
You can enter a transformation matrix or move the model manually: from the model icon menu select Transform coordinates and then choose one of the options..
The model is made the 'active' moving object which is indicated by a red cross on the left of the display table. To move the model you should hold down the X key and use either the left mouse button to rotate or the middle mouse button to translate the model.
In the window you can see the rotation matrix and translation vector that are currently applied to the coordinates and can reset these before clicking the Apply button to apply the transformation. Beware that you may need to click the Undo transformation button to undo any existing transformation. The program checks that your input has the correct number of elements and that the rotation matrix has a determinant of one.
This will return the model to its initial position.
The model will remain in the transformed position until the transformation is undone but the new coordinates will not be saved to file unless you explicitly do so. The transformation will be saved between program sessions and in status files and will be reapplied when the model is loaded. The new coordinates can be saved to file by selecting the File save/restore option from the model icon menu.
The Secondary Structure (SSM) superposition method superposes pairs of structures by the steps:
Find the secondary structure elements (SSEs) and represent them as one simple vector spanning
the length of the SSE
Find equivalent SSEs in the two structures using graph-theory matching by geometric criteria of distances and angles between the vectors
Superpose vectors representing equivalent SSEs
Find the most likely equivalent residues in the superposed SSEs
Superpose CA atoms of equivalent residues
Iterate the last two steps
The same procedure is also available on the EBI web server http://www.ebi.ac.uk/msd-srv/ssm
Krissinel & K. Hendrick (2004). Proceedings of CCP4 Study Weekend. Acta Cryst. D.
E.M. Mitchell et.al. (1989). J. Mol. Biol. 212 151-166