
|
CCP4 Molecular Graphics Tutorials | |
| Sequence Viewing and Alignment |
| Documentation Contents | On-line Documentation | Tutorials | CCP4mg Home |
Sequences for currently loaded coordinate files can be viewed by doing Applications->Sequence Viewer. A table will be displayed with a line for each chain in each coordinate file. One may also load in a set of sequences or alignment from a file in one of a variety of sequence formats (Clustal,EMBL,Fasta,Codata,AMPS,GCG/MSF,PIR,PFAM,SwissProt,Raw).
Hovering the mouse over a sequence entry will give information about the corresponding residue in the program status bar. Clicking on sequence entries will select/deselct them. Click-drag can be used to select and deselect ranges in a singles sequence or multiple sequences (try moving up and down with the mouse pressed down). Selecting sequence sections causes a graphical object with the corresponding atoms selected to be drawn in the graphics window, with a corresponding entry in the display table. An object is created for every entry in every chain the first that something is selected in the sequence viewer.The highlighted residues are drawn as "Fat bonds" by default, but like any other object, the style and colouring can be changed.
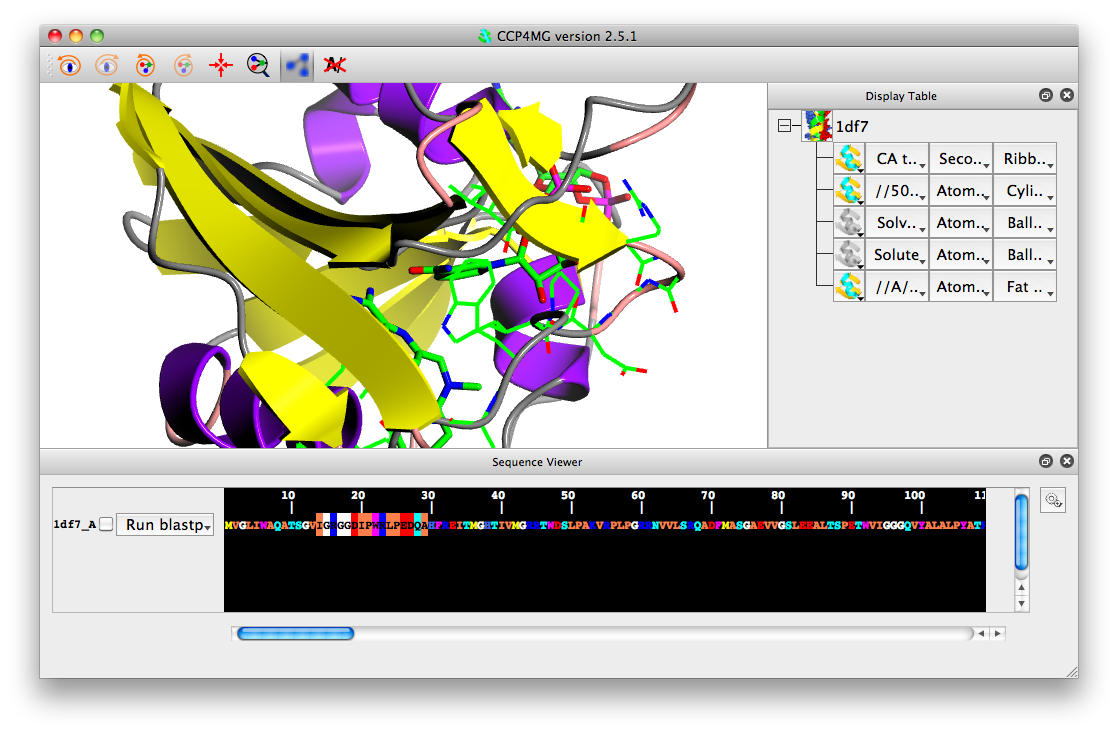
Load in a few coordinate files, e.g. 1df7, 4df4 and 8dfr. Select the check box beside the structure/chain name in the sequence viewer.
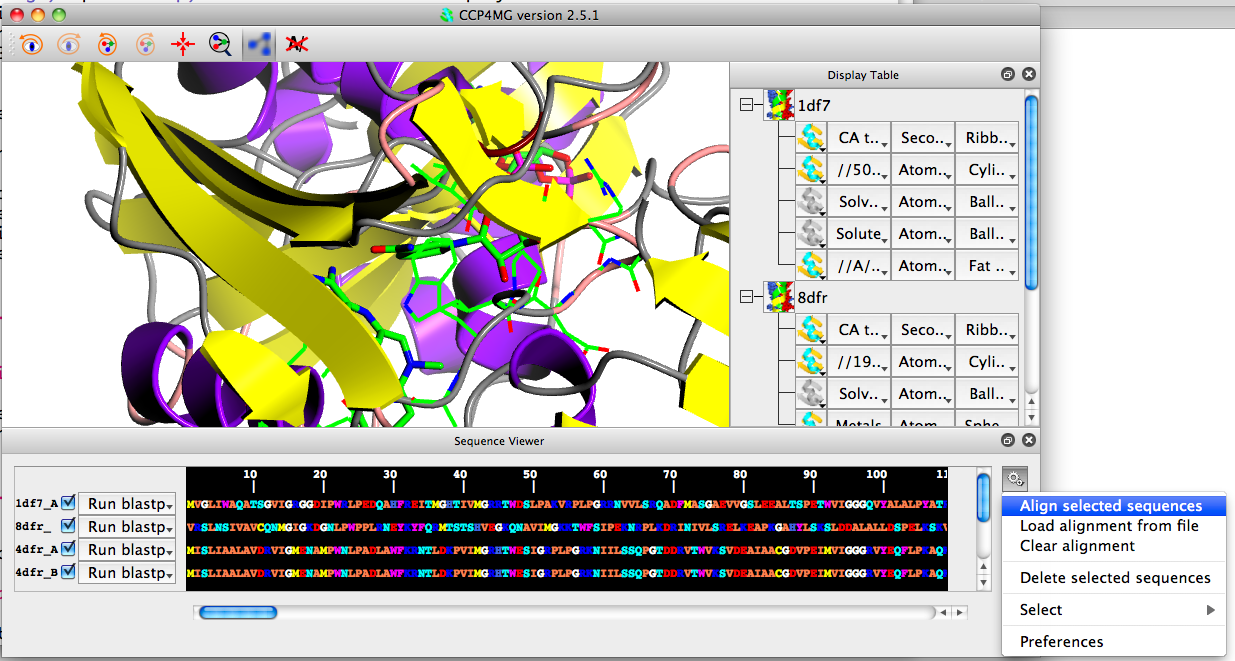
Click on the gear wheel icon at the right of the sequence viewer and click "Align selected sequences".
You should see that the sequences are now aligned and coloured be sequence conservation: green for fully conserved, yellow for partially conserved and red for non-conserved.
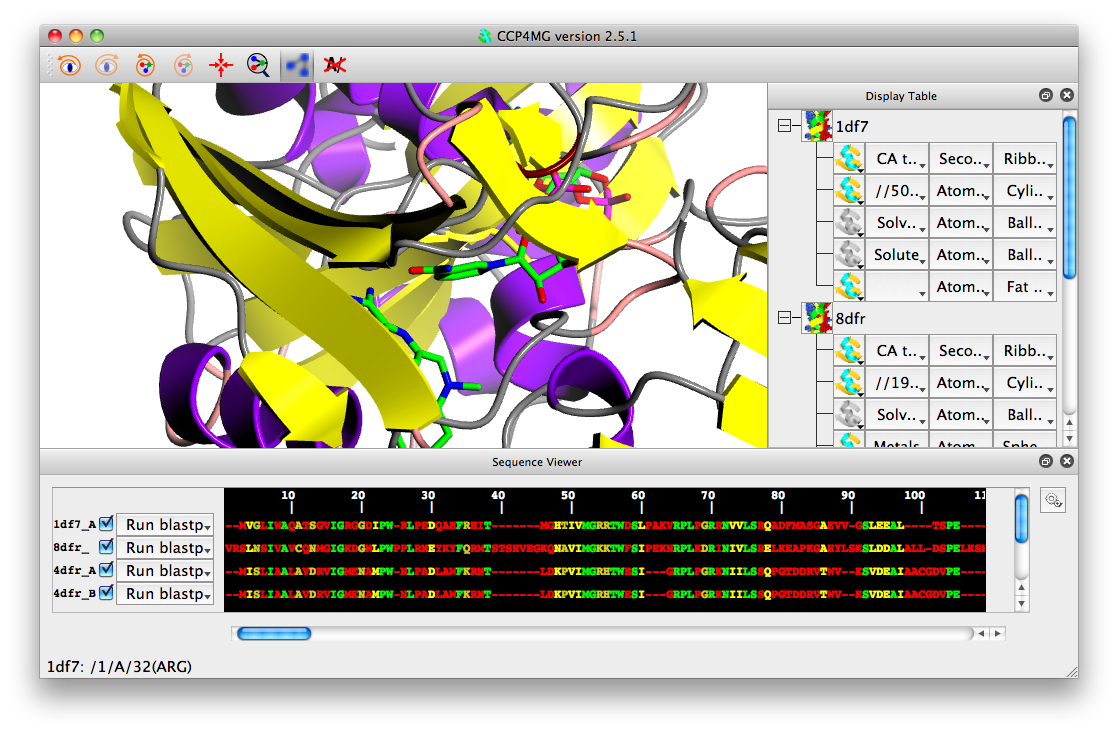
You can undo the alignment by selecting "Clear alignment" from the gear wheel icon.
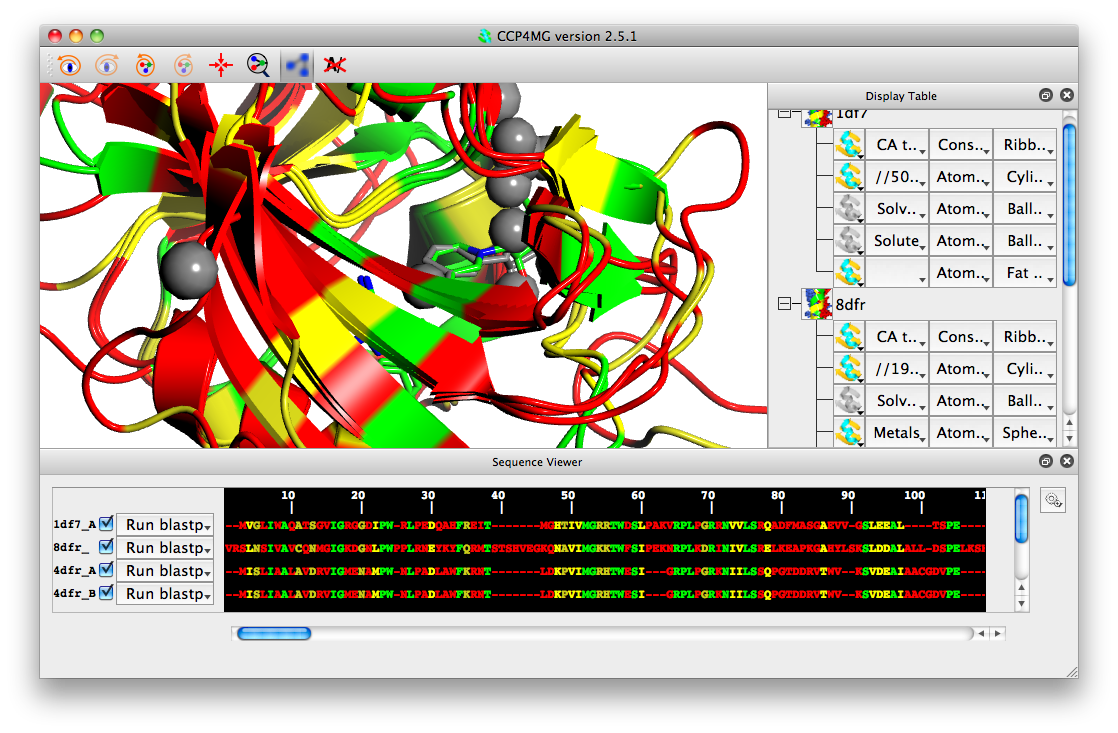
Once an alignment has been done you can colour models by sequence conservation. From the colouring menu for an object in the display table, select Residue property->Sequence conservation. If you also superimpose the structures (Applications->Superpose), then you should be able to produce an image like this:
It is possible to search for similar sequences (currently only amino acid) by using the NCBI blast tools. This can be done either by sending the target sequence to the EBI blast service or by using a local copy of the blast tools and database.Most users will not have access to a local copy of blast and will probably want to use the EBI option. However this is disabled by default: Sequence and structure information may be subject to an organization's data confidentiality regulations, a user may not have permission to send such information to an external service. This option is turned off by default to stop users accidentally using it. However, we have no such concerns with the tutorial data, so this option can be enabled.
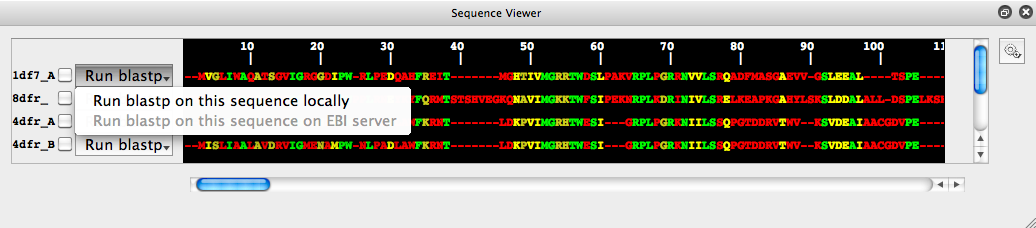
Click on the "Run blastp" button and then click on the "Run blastp on this sequence on EBI service" (even though grey, it can still be clicked). A box will pop-up, click on the edit-preferences in this box, check the "Allow running of blastp remotely" option, then click apply. The "Run blastp on this sequence on EBI service" option will be fully functional from now on and no longer greyed out.
From now on we can actually send our sequence to the EBI. Once again click on Run blastp->Run blastp on this sequence on EBI service. A confirmation window will appear, click on "Ok". The sequence will be sent to the EBI blastp web service to find similar sequences. This will take a little while! When this is completed, a window will appear listing structures with similar sequences and the score from blastp. You have the option to download some or all of these structures. A slider gives the option of automatically selecting structures based on blast score.
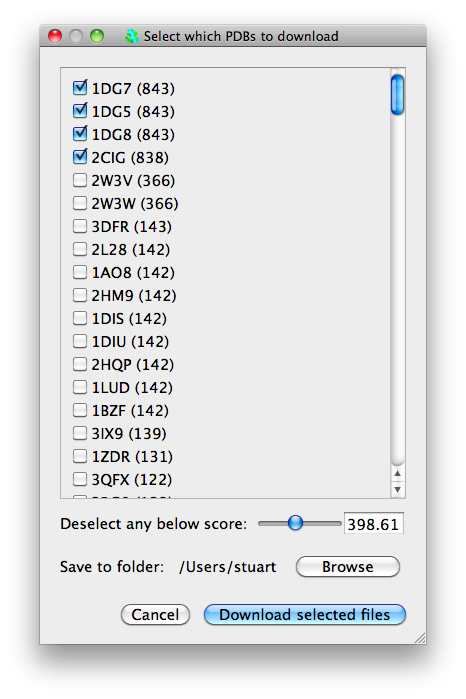
Select some structures (no more than about 5) and click "Download selected files". The structures will be downloaded and loaded into CCP4MG. However, they will not necessarily be structurally aligned nor will the sequences be aligned in the sequence viewer. (Future versions will apply the sequence alignment from blast and have option of structurally aligning based on the sequence alignment.)