1, Michail N. Isupov2
1CCP4, STFC Rutherford Appleton Laboratory, Research
Complex at Harwell, Harwell Science & Innovation Campus,
Didcot OX11 0FA, England
2Henry Wellcome Building for Biocatalysis,
College of Life and Environmental Sciences,
University of Exeter, Stocker Road, Exeter EX4 4QD, England
The presence of pseudosymmetry and especially its interplay with twinning may lead to an incorrect space group assignment. Moreover, if the pseudosymmetry is very close to an exact crystallographic symmetry, the structure can be solved and partially refined in the wrong space group. Typically in such false structures all or some of the pseudosymmetry operations are treated as crystallographic symmetry operations and vice versa. Such misassignment is not uncommon when the structure is solved by molecular replacement (MR) and it only becomes apparent, when the R-free ceases to decrease at about 35% or even at a higher value, and no further model rebuilding and refinement can improve it. At this point the electron density map remains imperfect (breaks in the main chain electron density, poor solvent peaks) while does not suggest any particular ways of model improvement.
The program Zanuda presented in this article was developed to automate the validation of space group assignment in such circumstances. In addition, the program can be used to restore the correct space group in structures which were intentionally solved in low symmetry space groups including P1. The validation is based on the results of a series of refinements in space groups, which are compatible with the observed unit cell parameters. Two assumptions are made in this method. Firstly, the pseudosymmetry operations, if any, are close enough to exact symmetry operations and, therefore, refinement converges to the global minimum when wrong symmetry constraints are removed and correct constraints are imposed. Secondly, the errors in individual macromolecules do not hinder the difference between pseudosymmetry and crystallographic symmetry, i.e. the model is already refined well enough (R-free around or below 40%). However it is not assumed that this refinement has been performed in the true space group.
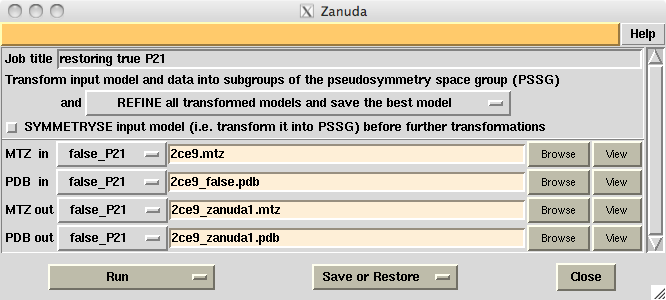
Zanuda is a Python script wrapping Refmac [1], several CCP4 [2] programs for handling MTZ-files and one purpose-written FORTRAN program for analysis of the pseudosymmetry in the input model and conversion of the model and data into possible space groups. Zanuda is included in CCP4 release 6.3.0. Its CCP4I task window (Fig. 1) can be opened from Validation & Deposition folder of the CCP4I GUI (task name "Validate space group") or from Program List folder (task name "Zanuda"). Zanuda summary file (Fig. 2) is explained further in the text. Originally the program was designed for YSBL server [3], where it runs in the default mode.
The program reads an input model and experimental data from files in PDB and MTZ formats, respectively. Both files are mandatory and must refer to the same space group and unit cell parameters. The input experimental data are to be presented as the observed structure amplitudes (not as intensities). Readability test is performed using Refmac in the mode of map coefficient calculation (zero cycles of restrained refinement). If the input files have not passed this test, the program stops and a user is prompted to correct or replace the input files and make sure that Refmac can read them.
The program has two modes. In the default mode it refines a series of models using Refmac and selects a model with highest symmetry from the ones with best refinement statistics. The program output includes this model in PDB format and a corresponding MTZ file from Refmac containing experimental data and map coefficients. Important, the transformed data in the output MTZ file are generated from already merged input data and should not be used at late stages of refinement; by no means can they be used for the PDB deposition. For these two purposes the experimental data are to be reprocessed accordingly.
One best structure is selected in this default automatic mode, while all the intermediate files are removed. However a refinement protocol and selection criterion universally suitable for all structures do not exist. Therefore, in the second mode of Zanuda no refinements are performed; instead all the transformed models and data are stored in a directory defined by a user. In the task interface, this mode can be activated using the drop-down list with the choice between "REFINE..." and "SAVE...". The second mode could be useful when, for example, refinement statistics for two structures are very similar and the automatic choice of one of them cannot be reliable. The initial models and transformed data for these two structures can be quickly obtained using this mode and then a more careful refinement and model rebuilding can be performed manually by a user.
Figure 1. CCP4I interface for Zanuda task with default settings.
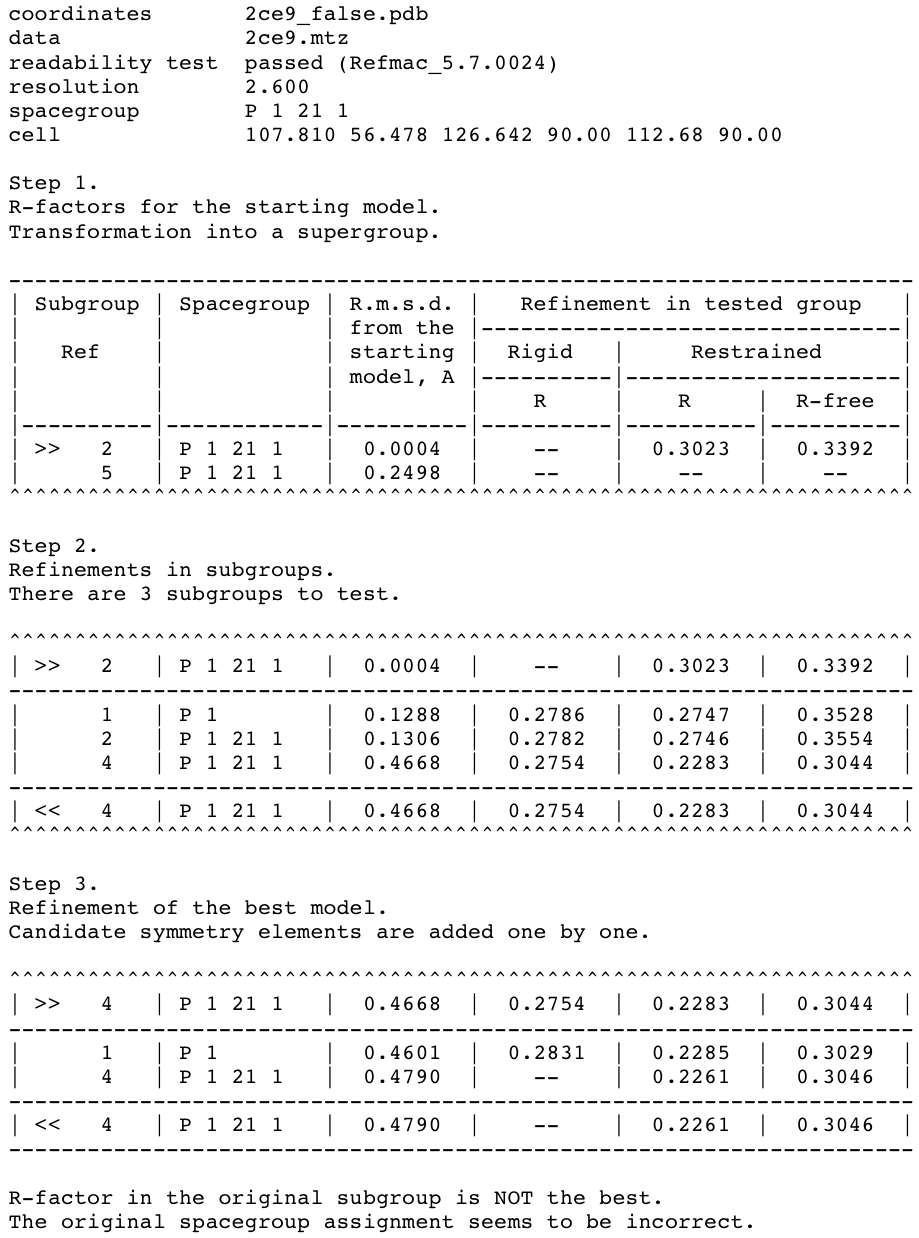
Figure 2. Correction of the space group assignment for the false origin structure generated from the PDB structure 2ce9.
This figure shows the summary file of Zanuda (with timestamps excluded). (Step 1) The input structure (subgroup 2) was transformed into the PSSG (subgroup 5) to calculate the r.m.s.d. of CA-atoms between the initial and symmetrised structures. (Step 2) The input structure was refined in candidate subgroups and (Step 3) transformed into the correct space group (subgroup 4). The input and output for a given step are marked by ">>" and "<<", respectively. All shown subgroups have equivalent unit cells except for the PSSG, which has the parameter a halved.
The space group of the crystal contains all the symmetry operations that map the crystal structure on itself. Similarly one can define a space group that, in addition, contains all the operations that perform approximate mapping, in a sense that the atomic coordinates need small adjustments to make the overlap between the structure and its copy exact. Such approximate operations are called pseudosymmetry operations and the extended space group will be further referred to as a pseudosymmetry space group (PSSG).
Noteworthy, the non-crystallographic symmetry (NCS) and pseudosymmetry are different concepts. An NCS operation is local and is defined by the best overlap of two NCS-related molecules after applying the NCS operation to one of them. On the contrary the pseudosymmetry operation is global and is defined by the best match between the entire crystal and its transformed copy. Thus the NCS operation and pseudosymmetry operation relating the same two molecules are in general different operations and may coincide only in special cases.
In structures with one molecule per asymmetric unit (AU) there is no pseudosymmetry and PSSG coincides with the space group of the crystal. In many cases of NCS, as, for example, in crystals with five molecules per AU, the global mapping of the crystal on itself cannot be defined even formally and PSSG remains equal to the crystal space group. In addition, Zanuda imposes the upper limit of 3 Å for the C-α r.m.s.d. between the structure and its copy generated by an additional global operation. Global operations with larger values of r.m.s.d. are ignored as they are unlikely to be misinterpreted.
Let us consider a structure with space group symmetry P21 and pseudotranslation vector a/2 (Fig. 3, Table 1). Its PSSG is a P21 space group with the basis of lattice vectors (a/2, b, c) (Fig. 3a,b). There are two P21 subgroups of the PSSG, both having the basis (a, b, c) compatible with the experimentally observed unit cell parameters. Let the first of these two subgroups be the true space group of the crystal structure (Fig. 3c,d). Then the second one is associated with the false origin structure in which pseudosymmetry axes are treated as crystallographic axes and vice versa (Fig. 3e,f). The two structures are different because different subsets of atoms are related by crystallographic symmetry, or, in other words, different symmetry constraints were implicitly imposed on the structures during refinement.
A coordinate file corresponding to the false structure (Fig. 3e,f) can easily be generated manually from the coordinates of the true structure (Fig. 3c,d) using, for example, the program Lsqkab from the CCP4 suite. At the start, the AU in the first structure may need to be redefined to include molecules related by pseudotranslation rather than by the rotation about a pseudosymmetry axis. Such an AU is convenient because it is also an AU in the second structure. Then the shift a/4 is applied to all atoms. (In a general case, the transformation from one subgroup to another is more complicated and includes, along with transformation of coordinates, extension of the AU or merging of several AUs together with averaging coordinates of related atoms.)
As a result of our transformation, the pseudosymmetry axes are treated as crystallographic axes (Fig. 3f). In the new model the relative positions of symmetry-related molecules (Fig. 3e), which are implicitly generated during refinement, are different from their relative positions in the true structure (Fig. 3c). Obvious consequences of such an error include bad refinement statistics and distorted electron density, usually with several breaks along the main chain.
False origin solutions are sometimes generated, when a structure with pseudotranslation is solved by MR. A real case of a false origin problem for a monoclinic structure was discussed in [4]. A more sophisticated example was encountered during MR structure solution of the GAF domain of CodY protein, PDB code 2gx5 [5]. The structure belonged to P4322 space group and had a pseudotranslation c/2. Therefore, three false origin structures were possible, one belonging to P4322 space group and two belonging to P4122. Another interesting example was reported in [6]. In this case the complete P31 structure was solved only after a false origin MR solution of P3121 substructure had been corrected.
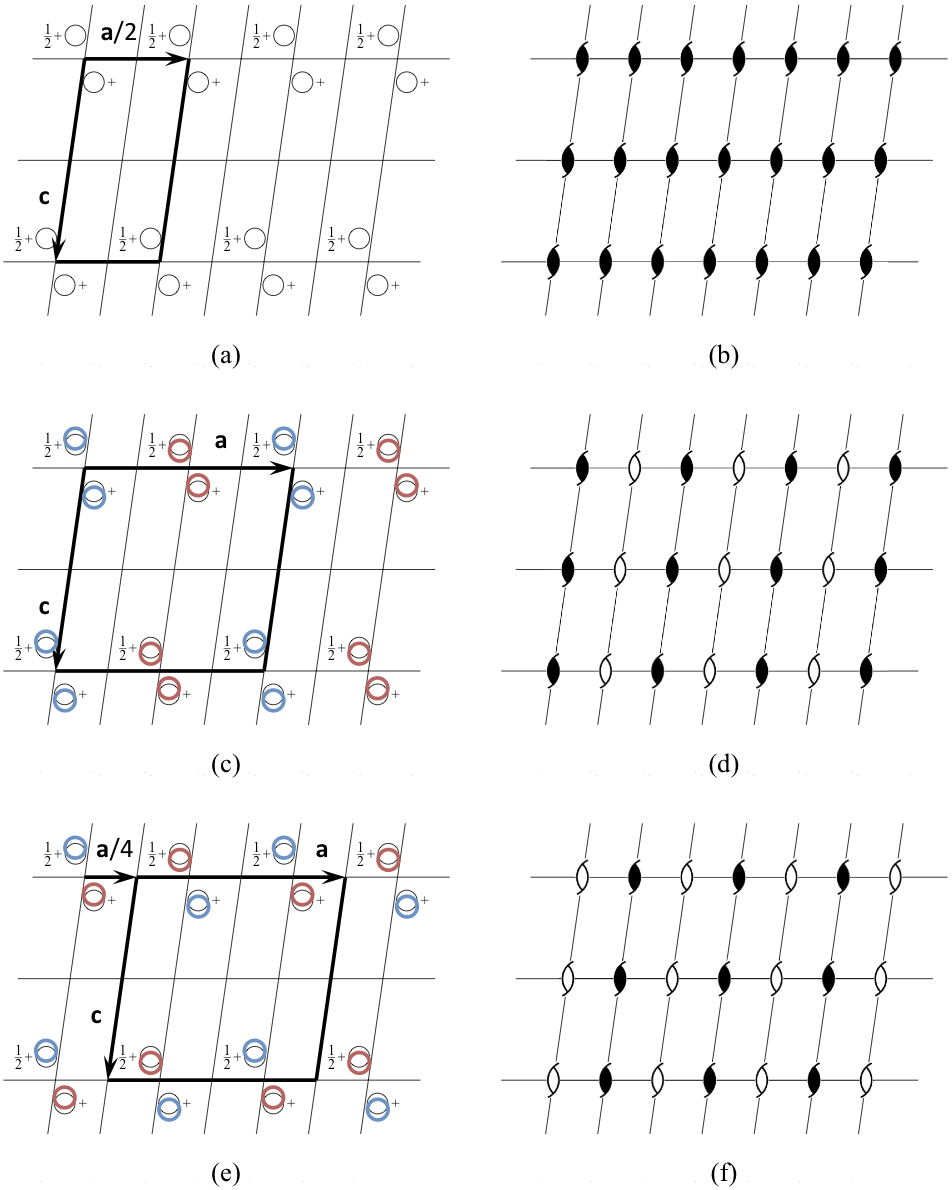
Figure 3. Pseudotranslation a/2 in a P21 space group.
Let the approximate structure (a, b), in which the pseudotranslation a/2 acts as crystallographic translation, be known. This structure belongs to space group P21 with the basis of lattice vectors (a/2, b, c). This leaves two possibilities for the true structure, (c, d) and (e, f). Both structures belong to the space group P21 with the basis of lattice vectors (a, b, c). The difference between the two structures is that crystallographic two-fold axes that are shown as filled shapes in (d) are treated as pseudosymmetry axes, which are shown as open shapes in (f), and vice versa. Accordingly, relative positions of symmetry related atoms, displayed as circles of the same colour in (c) and (e), are different. If, for example, (c, d) represent the true structure, then (e, f) represent an associated false origin structure. The standard crystallographic origin in P21 is located on the crystallographic two-fold axis, and, therefore, the crystallographic coordinates of corresponding atoms in the two structures differ by approximately a/4. In particular, this ambiguity may cause problems with structure solution using molecular replacement. Dependent on whether the first copy of search model is found at its true position or is displaced by a/4 from the true position, the molecular replacement will result in either the correct or the false origin solution.
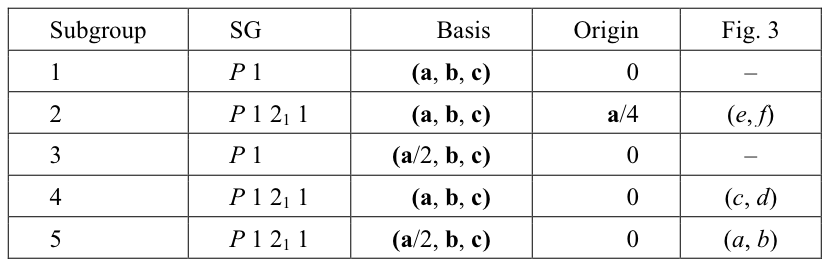
Table 1. Subgroups of the PSSG for a P21 structure with the pseudotranslation a/2.
Subgroup reference number used in summary file in Fig. 2 (Subgroup), space group Hermann-Mauguin symbol (SG), basis of lattice vectors (Basis), position of the standard origin relative to the standard origin in true structure (Origin) and references to the panels of Fig. 3 are shown for five subgroups of PSSG including PSSG itself. Among an infinite number of subgroups these subgroups have either smallest unit cells (3 and 5), or the basis of lattice vectors compatible with the experimentally observed unit cell parameters (1, 2 and 4).
The structure with PDB code 2ce9 [7] has the combination of symmetry and pseudosymmetry as shown in Fig. 3. The false origin structure can be generated from the true PDB structure as explained in the previous section, or using Zanuda in the no-refinement mode (selection "SAVE ..." in the mode list). The model generated using Zanuda was further refined with strong geometrical restraints and then used as an input for the demonstrative Zanuda run presented in Figs. 1 and 2.
Summary file (Fig. 2) contains a description of the input (including the confirmation that the input files have passed the readability test) and three tables corresponding to three steps of Zanuda protocol. Each table row corresponds to an atomic model. A reference number of a subgroup of PSGG to which the model belongs is given in the first column. The first column may also contain a symbol denoting the role of the model for this step, with ">>" and "<<" standing for input and output models, respectively. The space group Hermann-Mauguin symbol for the subgroup is shown in the second column. The reference numbers and space group symbols for relevant subgroups are also listed in Table 1.
The third column shows C-α r.m.s.d. of a given model from the input model. This is a global deviation between two infinite crystal structures, not between two AUs or two molecules. For example, this number for the final model is small, of order of 0.1 Å, if the initial and final subgroups are the same. On the contrary, if the initial model was substantially incorrect (pseudosymmetry operations were treated as crystallographic operations), then the r.m.s.d. for the final model will be larger, typically 0.5 to 2 Å.
The last three columns present R-work after rigid body refinement, and R-work and R-free after restrained refinement. However, at the Step 1 of the protocol no refinement is performed and the only two R-factors shown are for the modified input model (see the next paragraph). Also, rigid body refinement at Step 3 is performed only for the P1 model and remaining rigid-body R-factor columns are void. In the no-refinement mode all the R-factor columns are void.
At Step 1, the PSSG is determined, the input model is modified and transformed into PSSG. The modification involves the removal of solvent and of those residues, which have no match in at least one of the pseudosymmetry related chains. The first row in the table corresponds to the modified model in the original subgroup. The second row corresponds to the model transformed into PSSG. In the default mode discussed in our example, this transformed model is not used further on and is shown for information only. R.m.s.d. for this structure characterises the deviation of the pseudosymmetry in the input structure from the exact crystallographic symmetry. Limiting value here is 3 Å; larger deviations of pseudosymmetry operations from exact symmetry do not usually hinder the space group assignment and are not included in PSSG.
Step 2 involves independent refinements in those subgroups of the PSSG, which have the basis of lattice vectors compatible with observed unit cell parameters. In our example this criterion was satisfied for one P1 subgroup (subgroup 1 in Fig. 2 and Table 1) and two P21 subgroups (subgroups 2 and 4). After this step it was already quite clear that the subgroup 4 was the correct one. The structure refined in this subgroup deviated from the input model significantly more (r.m.s.d. 0.47 Å) than structures in subgroups 1 and 2 (r.m.s.d. 0.13 Å), and it was a change in the right direction as indicated by substantially lower R-free for this model (30.4%) compared to the other two (35.3% and 35.5%). One could have expected that refinement in P1 (subgroup 1) would also be able to improve the model because there are no rotational-symmetry constraints in this space group. However, this has not happened because the previous refinement in an incorrect subgroup (during the input model preparation) pushed the model into a wrong local minimum from which the model can not escape in the course of refinement in P1.
The model with lowest R-free is passed to Step 3, where it is expanded to P1 (subgroup 1), refined, and then symmetry operations are added one by one, with a round of refinement after each addition. In our case this procedure only confirms that the subgroup 4 is the right answer, but in general comparison of R-free factors after these refinements provides a reasonable criterion for final subgroup selection. In fact this procedure alone would have been sufficient, provided the refinement in P1 always converge to a correct minimum. However this is not always the case – as demonstrated by our example – and series of refinements in subgroups at Step 2 give more chance of a successful structure correction.
In more detail, the following actions are performed at the Step 3. Firstly, rigid body and restrained refinement are carried out for the input model extended to the space group P1 (subgroup 1). This is followed by one or more cycles of increasing the symmetry. At each cycle an attempt is made to find such a symmetry operation from PSSG that (i) hasn't been used in previous cycles, and (ii) does not result in changes of primitive unit cell parameters - so the pseudotranslation operations are never added. If several non-equivalent operations are available, the program chooses the one, which gives a minimal r.m.s.d. between the previous refined structure and its copy generated using the operation under consideration. If found, such a symmetry operation and the space group obtained in the previous cycle define a new space group with higher symmetry; the refined model from the previous cycle is transformed into the new space group and refined again. Only if the new operation is found and refinement in the new space group has lead to a decrease in R-free or to its increase by no more than 2%, Zanuda moves to a new cycle and the described procedure is repeated. Otherwise it terminates and outputs the result from the previous cycle.
In our example two such cycles were completed. At the first cycle, a screw two-fold rotation operation was added resulting in the space group P21 (subgroup 4). After refinement in this space group, the increase in R-free was substantially less than the threshold value, so the second cycle started. However, the remaining symmetry operations included pseudotranslations and rotations about the screw two-fold pseudosymmetry axes, and each of them would extend the subgroup 4 to the complete PSSG (subgroup 5) with a smaller unit cell. Therefore, the second cycle was terminated and the structure refined in subgroup 4 was accepted as a final result (Fig. 2).
If a symmetry operation is missing in the space group from which the model is transformed but is present in the space group to which the model is transformed, then coordinates of atoms related by this operation are averaged and atoms are merged. Of course no such averaging happens for the models representing the initial space group and its subgroups including P1. Therefore these models could remain strongly biased towards incorrect symmetry and refinement alone may be unable to pull these models out of the local minimum associated with the incorrect symmetry. This is exactly what has been observed in our example during P1 refinement at step 2.
Preliminary transformation of the model into PSSG (check box "SYMMETRISE ...") helps the model escape from such local minima. In addition, with this approach all the refinements at Step 2 start, in effect, from the same crystal structure, which allows more strict comparison of subsequent refinements. If this option were used in our example, then the correct symmetry would have been restored after refinements in both the correct space group P21 (subgroup 4) and space group P1 (subgroup 1).
Preliminary transformation into PSSG helps reduce the bias of the model towards incorrect symmetry, but still does not guarantee a success. Probably a more efficient method would be to start from the structure solved in P1 by MR. Then the true space group can be recovered using Zanuda. (The option "SYMMETRISE ..." should not be used here). The drawback of such approach is that it might prove difficult if not impossible to solve the structure in P1 by MR using the original search model. An attempt to sidestep this difficulty by using the search model refined in the initial, incorrect subgroup, may again result in an incorrect structure.
[1] Murshudov, G. N., Skubák, P., Lebedev, A. A., Pannu, N. S., Steiner, R. A., Nicholls, R. A., Winn, M. D., Long, F. & Vagin, A. A. (2011). Acta Cryst., D67, 355–367.
[2] Winn, M. D. et al. (2011). Acta Cryst., D67, 235–242.
[3] http://www.ysbl.york.ac.uk/YSBLPrograms/
[4] Isupov, M. N. & Lebedev, A. A. (2008). Acta Cryst., D64, 90–98.
[5] Levdikov, V. M., Blagova, E., Colledge, V. L., Lebedev, A. A., Williamson, D. C., Sonenshein, A. L. & Wilkinson, A. J. (2009). J. Mol. Biol., 390, 1007–1018.
[6] Watson, A. A., Lebedev, A. A., Hall, B. A., Fenton-May, A., Vagin, A. A., Dejnirattisai, W., Felce, J., Mongkolsapaya, J., Screaton, G. R., Murshudov, G. N. & O'Callaghan, C. A. (2011). J. Biol. Chem., 286, 24208–24218.
[7] Jennings, B. H., Pickles, L. M., Wainwright, S. M., Roe, S. M., Pearl, L. H. & Ish-Horowicz, D. (2006). Mol. Cell, 22, 645–655