RAPPER:
Real Space Automated Conformer Generation
Nicholas Furnham‡ and Tom L. Blundell
Department of Biochemistry, Cambridge University
80 Tennis Court Road, Cambridge, CB2 1GA, UK
‡ Corresponding Author: nick@cryst.bioc.cam.ac.uk
Overview
Generating protein conformers by discrete sampling of likely conformers within a given set of restraints provides an attractive method for answering a number of important questions in structural biology [de Bakker, et al., 2006]. This approach, now incorporated in RAPPER, allows the generation of solutions consistent with a combination of restraints derived from a wide variety of sources, both experimental and theoretical. RAPPER can be applied to a number of problems including ab initio loop building [de Bakker, et al., 2003], comparative modelling [Furnham, et al., 2007] and Cα-trace modelling [DePristo, et al., 2003]. It can also be used to build and refine conformers using X-ray crystallographic data [DePristo, et al., 2004, DePristo, et al., 2005, Furnham, et al., 2006], and this is now being integrated into the CCP4 software suite. The analysis tool RAMPAGE [Lovell, et al., 2003] is also incorporated into RAPPER in order to allow examination of j / y values using the most up-to-date propensities to generate a Ramachandran diagram.
Methodology
At the heart of RAPPER lie two principal engines: restraint generation and fast and efficient sampling. The latter uses idealised geometry as the model representation of a protein (as formulated by Engh and Huber [Engh and Huber, 1991]) to generate conformers of main chain and, optionally, side chain heavy atoms. By using idealised geometry the number of degrees of freedom can be reduced to the major degrees of freedom present in a protein (j, y, w and c). RAPPER samples j / y space using fine grained (5° x 5°) residue-specific propensity-weighted maps derived from the Top500 database of protein structures [Lovell, et al., 2003]. The w dihedral angle is sampled as either the cis or trans state with the prevalence observed in the PDB [Berman, et al., 2000]. Special consideration is given to pre-proline residues which have a higher propensity to occur in the cis state [Jabs, et al., 1999]. In addition to this, side chain modelling is performed by sampling a rotamer library [Shetty, et al., 2003]. Although any library can be used, the principal library employed by RAPPER is the ‘penultimate’ library, which is derived in a similar way to the j / y maps [Lovell, et al., 2003].
The sampling algorithm itself is based on a ‘branch and bound population search’ procedure. Essentially this comprises a sequential directed search from N to C termini, though there is no fundamental limitation to this and it equally could be C to N. At each residue a population of conformers (children) that extend the previous set of conformers (parents) that satisfy all the enforced restraints are searched. At each level in the search, pruning of an otherwise ever expanding search tree is achieved by maintaining only the conformers that satisfy the current restraints. The result is an exponential decrease in the number of conformers. In practice the size of the population is fixed at 100 and the number of iterations permitted to generate the child population is 100,000. This is summarised in Figure 1.
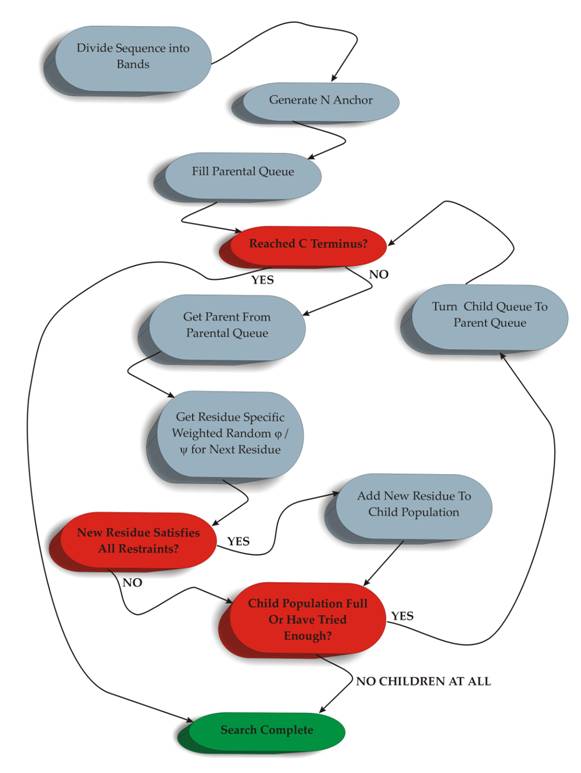
Figure 1: Overview of the RAPPER Sampling Algorithm. Decision points are shaded in red and completion in green.
Sampling occurs within a given set of restraints. These can be both problem-specific and general. Common to all modelling is the hard-sphere excluded-volume restraint. This enforces a minimum distance between atoms, the van der Waals radii for which are taken from PROBE [Lovell, et al., 2000]. The process of checking for these excluded volumes can be very computationally expensive. This problem is addressed in RAPPER by an efficient grid-based checking system, which holds only a small substructure of atoms at each grid point that might overlap with an atom lying near that point. The number of atoms at the grid point that are required to be checked is dependent only on the resolution of the grid and the distribution of the fixed atoms and is independent of the size of the protein. In addition the rate at which atoms are fixed to the grid as they are being built is optimised to a single common ancestor at most twenty residues preceding the residue being built. Thus only a small fraction of the diverse population local (in sequence space) to the residue being built needs to be checked; the rest are reduced to a single shared path of the search tree. Problem-specific restraints include positional restraints to ensure that the sampled conformers fall within a specific position in space; this is utilised in tracing a structure from Cα-atom positions. Other restraints derived for loop modelling include gap closure, C-terminal anchor dihedral angle minimisation and strict C-anchor positioning. For X-ray crystallography electron density can be used to derive restraints by ensuring that atoms from the sampled conformers lie within electron density cut-off by a minimum σ threshold.
Applications
The generalised nature of the sampling algorithm permits the application of RAPPER to a number of problems in structural biology. In the first instance it was applied to generating loop regions [de Bakker, et al., 2003]. Further development permitted the regeneration of the entire protein from Cα-atoms [DePristo, et al., 2003]. Most recently the combination of these early developments with restraints from electron density has assisted X-ray structure determination.
Although there has been significant progress in developing model building and refinement techniques for structure determination by X-ray crystallography, it still remains a challenge to produce reliable high quality models of proteins from medium and low resolution crystals. This is primarily due to the difficulty in adequately exploring the large and complex energy landscape and determining the set of conformers that best describes the experimental data. In order to address this problem we have explored the use of the real space knowledge-based conformational sampling in RAPPER coupled with reciprocal space refinement by either molecular dynamics / simulated annealing (MD/SA) or maximum likelihood.
We have demonstrated that the combined approach of RAPPER and MD/SA greatly improves the quality and power of refinement when compared to MD/SA alone at medium resolution (2.0-3.0Å) [DePristo, et al., 2004, DePristo, et al., 2005]. This is shown through the re-determination of two previously solved structures, providing a controlled situation to test the procedure, as well as a more realistic challenge of a blind test structure determination. In comparing the combined approach to MD/SA alone we show that RAPPER is able, by discrete sampling, to cross deep wells within the X-ray potential energy landscape from which MD/SA alone cannot escape, particularly those involving bulky side chains. Limiting the degrees of freedom by sampling only good j/y characteristics or rotameric side chains and the enforcement of prior values, such as Engh-Huber bond angles and lengths gained from knowledge of protein structure, avoids exploring unlikely and fruitless areas of conformational space. It is these areas that, at lower resolutions, are likely to accommodate conformations with poor j/y and rotameric side chains. This approach helps to overcome the limited specificity, modal bias and phase error of lower resolution maps.
The approach has been further developed to push the boundaries of de novo model building and refinement to lower resolutions (>3Å). This has been demonstrated through the application of RAPPER, along with refinement by maximum likelihood, to the structure of Lif1-Lig4, a 90KDa complex of two protomers involved in the final ligation step of DNA non-homologous end joining. The native dataset from crystals of this complex was collected to 3.9Å resolution, and experimental phases were collected to ~4.5Å resolution [Dore, et al., 2006].
The advantage of using RAPPER to model into low resolution data is that it enables the testing of weak hypotheses, assumptions and often speculations about structures suggested by the electron density map. Sequence registry indicated by features in the density as well as information inferred from homologous sequences and structures can be tested. Efficient exploration of conformational space allows the uncertainty of the dataset to be taken into account; this is important for low resolution data [Furnham, et al., 2006].
In all applications RAPPER is able to generate an ensemble of solutions that more or less equally fit the data. Such a crystallographic ensemble can represent one of two possibilities. Firstly, there may be several individual structures that are equally compatible with the data when considered one at a time; these can be thought of as describing the uncertainty of the experimental data. Secondly it can represent spatial heterogeneity and dynamics: a set of structures that, taken over time and the different unit cells, are compatible with the data. Obviously such ensembles can reflect aspects of both factors and at present we are unable to distinguish between the two, but in any case the collection epitomizes the inappropriateness of specification of a model as a single species.
RAMPAGE
The use of the fine grained j / y residue specific propensity-weighted maps derived from the Top500 database of protein structures in RAPPER allows for the geometrical evaluation of a protein model to be undertaken. To this end RAMAPGE [Lovell, et al., 2003] has been developed in collaboration with the Richardson Group. This generates a Ramachandran diagram plotting j verses y backbone conformational angles for each residue in the model. These can be classified into regions of favoured, allowed and disallowed regions. The contouring of these regions is based on density-dependent smoothing for 81,234 non-Gly, non-Pro, and non-prePro residues with B < 30 from 500 high-resolution proteins. Delineation between large empty areas and regions that are allowed but disfavored can be discerned. One such region is the g-turn conformation near +75°,-60°, counted as forbidden by common structure-validation programs; however, it occurs in well ordered parts of good structures and is overrepresented near functional sites. Regions are also defined for Pro, pre-Pro, and Gly (see Figure 2 and 3). These plots provide crystallographers with an up to date tool for locating and fixing local problems during the process fitting electron density and refinement as well as a general assessment of model quality and the clarify if a structure generally meets current standards of good practice in the field [Lovell, et al., 2003].
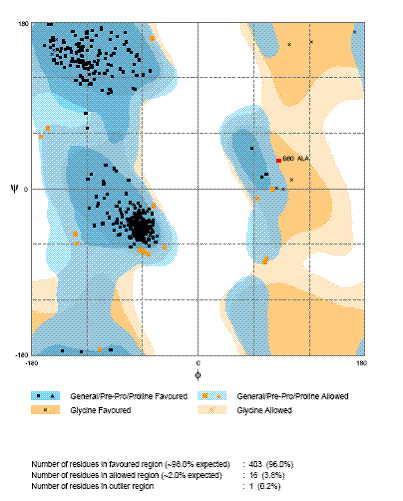
Figure 2: Ramachandran Plot Generated by RAMAPGE. The j / y distribution, as analysed by RAMPAGE, for the structure of the complex Xrcc4 and a fragment of Ligase IV (PDB code 1IK9).
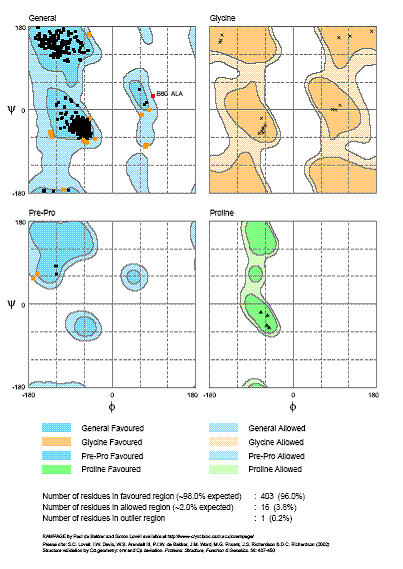
Figure 3: General, Glycine, Pre-Proline and Proline Specific Ramachandran Plots Generated by RAMPAGE. The j / y distribution, as analysed by RAMPAGE, for the structure of the complex Xrcc4 and a fragment of Ligase IV (PDB code 1IK9). The specific propensities for Glycine, Pre-Proline and Proline are analysed in separate Ramachandran plots.
RAPPER & CCP4
As part of the CCP4 suite of programs, RAPPER is intended to provide a useful conformer generation tool to be used at a number of points in the model building process. Partial models can be rebuilt correcting for sequence assignment and positioning of residues generated by molecular replacement (MR) or extending the model between structural elements generated by automated building programs such as Buccaneer [Cowtan, 2006]. Once a complete or near complete model has been generated RAPPER, in combination with a refinement programme such as REFAMC, can be used to identify automatically, and rebuild poor fitting regions before refining the structure. In a more user interactive mode of operation, where weak or difficult to interpret density exists (both at reasonable resolution and low resolution), RAPPER can be a powerful tool in providing sets of conformers consistent with the experimental data and taking into account any prior knowledge about the structure. To aid the entire process it is hoped that RAPPER will be able to be used though COOT [Emsley and Cowtan, 2004]. Further information can be found at the RAPPER website http://mordred.bioc.cam.ac.uk/~rapper/.
References
1. Berman, H.M., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T.N., Weissig, H., Shindyalov, I.N., and Bourne, P.E. (2000). The Protein Data Bank. Nucleic Acids Res 28, 235-242.
2. Cowtan, K. (2006). The Buccaneer software for automated model building. 1. Tracing protein chains. Acta Crystallogr D Biol Crystallogr 62, 1002-1011.
3. de Bakker, P.I., DePristo, M.A., Burke, D.F., and Blundell, T.L. (2003). Ab initio construction of polypeptide fragments: Accuracy of loop decoy discrimination by an all-atom statistical potential and the AMBER force field with the Generalized Born solvation model. Proteins 51, 21-40.
4. de Bakker, P.I., Furnham, N., Blundell, T.L., and Depristo, M.A. (2006). Conformer generation under restraints. Curr Opin Struct Biol.
5. DePristo, M.A., de Bakker, P.I., and Blundell, T.L. (2004). Heterogeneity and inaccuracy in protein structures solved by x-ray crystallography. Structure (Camb) 12, 831-838.
6. DePristo, M.A., de Bakker, P.I., Johnson, R.J., and Blundell, T.L. (2005). Crystallographic refinement by knowledge-based exploration of complex energy landscapes. Structure (Camb) 13, 1311-1319.
7. DePristo, M.A., De Bakker, P.I., Shetty, R.P., and Blundell, T.L. (2003). Discrete restraint-based protein modeling and the Calpha-trace problem. Protein Sci 12, 2032-2046.
8. Dore, A.S., Furnham, N., Davies, O.R., Sibanda, B.L., Chirgadze, D.Y., Jackson, S.P., Pellegrini, L., and Blundell, T.L. (2006). Structure of an Xrcc4-DNA ligase IV yeast ortholog complex reveals a novel BRCT interaction mode. DNA Repair (Amst) 5, 362-368.
9. Emsley, P., and Cowtan, K. (2004). Coot: model-building tools for molecular graphics. Acta Crystallographica Section D-Biological Crystallography 60, 2126-2132.
10. Engh, R.A., and Huber, R. (1991). Accurate Bond and Angle Parameters for X-Ray Protein-Structure Refinement. Acta Crystallographica Section A 47, 392-400.
11. Furnham, N., de Bakker, P.I., Gore, S., Burke, D.F., and Blundell, T.L. (2007). Comparative Modelling by Restraint-based Conformational Sampling
12. Furnham, N., Dore, A.S., Chirgadze, D.Y., de Bakker, P.I., Depristo, M.A., and Blundell, T.L. (2006). Knowledge-based real-space explorations for low-resolution structure determination. Structure 14, 1313-1320.
13. Jabs, A., Weiss, M.S., and Hilgenfeld, R. (1999). Non-proline cis peptide bonds in proteins. J Mol Biol 286, 291-304.
14. Lovell, S.C., Davis, I.W., Adrendall, W.B., de Bakker, P.I.W., Word, J.M., Prisant, M.G., Richardson, J.S., and Richardson, D.C. (2003). Structure validation by C alpha geometry: phi,psi and C beta deviation. Proteins-Structure Function and Genetics 50, 437-450.
15. Lovell, S.C., Word, J.M., Richardson, J.S., and Richardson, D.C. (2000). The penultimate rotamer library. Proteins 40, 389-408.
16. Shetty, R.P., De Bakker, P.I., DePristo, M.A., and Blundell, T.L. (2003). Advantages of fine-grained side chain conformer libraries. Protein Eng 16, 963-969.