Multiple rotation function
By L. Urzhumtseva & A. Urzhumtsev
Laboratory of Crystallography and Modelling of
Mineral and Biological Materials, UPRESA 7036 CNRS, University Henri Poincaré,
Nancy I, 54506 Vandoeuvre-les-Nancy, France
e-mail :
sacha@lcm3b.uhp-nancy.fr
Introduction
One of principal tools of macromolecular crystallography, the molecular replacement procedure (Rossmann, 1972, 1990), is based on several assumptions the main of which is the following :
- the search model is sufficiently close to the model of the crystal under study (or to its large enough part) so that it fits best to the experimental structure factor magnitudes being placed at the correct position.
While the search directly in the 6-dimensional space is eventually possible, specially with modern computers and efficient algorithms (Chang & Lewis, 1997; Kissinger et al., 1999), it does not solve the problem when the model is imperfect and the Main Assumption is not verified. In such difficult cases, two separate consecutive searches in three-dimensional spaces, rotation and translation, can have an advantage.
The rotation search is traditionally done by comparison of Patterson maps, when even a partial model eventually can be recognised. More difficulties arise for search models composed from several blocks whose relative orientation is different from that in the molecule under study. For a small number of such rigid groups, a so-called PC refinement (Brünger, 1990; DeLano & Brünger, 1994) can help weaking therefore the Main Assumption.
At the step of the translation function, traditionally the structure factor magnitudes calculated from the search model are compared with the corresponding experimental values. If the search model is quite incomplete or contains significant errors, there is no many reasons why this fit will be best when the model is placed correctly (a long history of Molecular Replacement shows many examples pro and contra). Often, a posteriori analysis shows that the solution was in the list but it was difficult to recognise it among a very large number of possible positions. In this case, the knowledge of the model orientation could remove many spurious peaks for wrong orientations and thus solve or simplify the problem.
Alternatively, a search with incomplete models can be improved when maximum likelihood (ML) approach (Read, 1999, communication at the IUCr Meeting, Glasgow) is used. This technique allows to take into account a missing part of the model (note that here the Main Assumption changes its original form; the calculated structure factors are not fitted directly to the experimental values; some discussion can been found, for example, in Lunin & Urzhumtsev, 1999). However, the ML criterion is essentially more time consuming, there is no evident way to calculate it rapidly as it is done for the least-squares criteria (Navaza, 1994; Navaza & Vernoslova, 1995). In this case, the reduction of the number of possible orientations from several tens to a few possible variants is also crucial.
Multiple rotation function analysis
When the molecular replacement
search does not give an evident solution, an old idea is to repeat the search
varying the models (whole model, main chain model, Ca model, a model with deleted
loops, etc), the set of structure factors (for example, selected by its
resolution) or the parameters (for example, the integration radius). If no
evident solution appears, many rotation functions are analysed together with
the hope that the signal is consisted and can be identified in many of these
functions. Such comparative analysis is not at all transparent because it is
complicated to estimate visually the closeness of rotation angles, specially
when the space group has symmetry operations and when the programs like AMoRe
(Navaza, 1994) make some pre-rotation of the model before the search.
Such a comparison of several rotation functions is important also when
the search is done with NMR models. Usually, they are several tens, neither of
them is quite close to the correct model, rotation functions are quite noisy
with the correct answer hidden in the middle of the list of peaks.
The goal of our current approach is to find unambiguously the molecular
orientation in the crystal. With this, the Main Assumption can be replaced by a
weaker one :
- a model taken in its correct orientation fits well enough to the experimental data in comparison with all its other positions in the same orientation
Computationally,
the knowledge of the orientation (or a few orientations) allows to test
possible positions with more sophisticated, powerful but time-consuming
criteria, take into account all structure factor corrections like the bulk
solvent correction etc. This article does not concern the study of this improved
translation searches which is the object of our independent work and deals only
with the rotation analysis of many rotation functions considered
simultaneously.
In order to compare several rotation functions, the following procedure has been proposed :
1)
Rotation functions are calculated varying the
models and/or parameters of the
rotation function including the resolution of the data set; if several search
models are tested, they must be superimposed before to calculate the rotation
functions ;
2)
For each pair of the rotation angle triplets (am, bm, gm) and (an, bn, gn) coming from all lists of
the peaks, the distance between them is calculated taking symmetry operations
into account ;
3)
A clustering procedure is applied for the
calculated matrix of distances; the clustering results are represented in the
form of a cluster tree and the clusters are defined varying the minimal
interangular distance; for a chosen cut-off level of the interangular distance,
the peaks inside the cluster are considered to be coincided, the size of all
clusters is calculated and used as the information to choose the solution.
We believed that such procedure will give a signal because noisy peaks are distributed relatively randomly in the space and therefore are associated to different clusters while the correct peaks should be close enough each to others and will belong to the same cluster. Moreover, there is the second reason. Usual variations in the arrangement of secondary structure elements will lead to several optimal orientations of the same model relatively close each to other – in one orientation one group of the secondary structure elements is superimposed better, in another orientation – another group.
Several comments can be done.
First, while in our work all molecular replacement searches where done
by AMoRe (Navaza, 1994), the analysis of the rotation function is general and
can be applied to lists of rotation function peaks obtained by any means but
expressed in Eulerian angles a, b, g (see Urzhumtseva &
Urzhumtsev, 1997, for different rotation systems). The peak comparison is done
for the final values of the rotation angles; this means that for the
programs like AMoRe that preliminary puts the model to some special
orientation, the lists of peaks or1.s are compared not directly but
using corresponding files tabl1.s for the pre-rotations. The program
gives the answer in both terms.
Second, the distance between a pair of rotation angles is expressed
through the effective rotation angle k between two
corresponding model orientations. If Mm and Mn are corresponding rotation matrices then the
matrix of the relative rotation is calculated as the product MmMn-1 and the corresponding
efficient rotation angle is calculated as
k = arccos{ [trace(MmMn-1) – 1] /2 }.
If
the space group contains several symmetry operations, the distance is chosen as
the minimal value of distance calculated for all symmetry related pairs.
Distance between two clusters is defined as the minimal distance between all pairs
of rotation angles, one from each cluster. When a noncrystallographic rotation
presents in the crystal and its order and the axis direction are known from the
self-rotation function, this operation can be also considered at the step of
the distance calculation allowing to identify the pairs of angle triplets
linked by this symmetry and to enforce the signal. Various distances can be
defined for a given pair of angles. However, the architecture of the cluster
tree will be the same for any of these definitions as soon as the distance
increases with the effective rotation angle which seems to be logical.
Third, when the size of a cluster is calculated, the coincidence (or
closeness) of higher peaks could cost more that the coincidence of lower peaks;
therefore, the contribution of every rotation function peak can be weighted,
for example, by its height. This can be interpreted as an integral measure of
the peaks coincidence. The level at which rotation angles are considered to be
coincided and the cluster size is calculated cannot be defined once forever. It
is an important parameter of an interactive search of the answer.
The suggested procedure was realised in a FORTRAN program with an
interactive interface in Tcl/tl (Ousterhaut, 1993). This program allows to read
a list of rotation function files (or1.s in AMoRe format) and
corresponding pre-orientation protocols (tabl1.s in AMoRe format), to
define a list of symmetry operations including noncrystallographic symmetries
if available, to obtain a cluster tree with references to the initial rotation
functions, to define the cluster size with a variable cut-off level of the
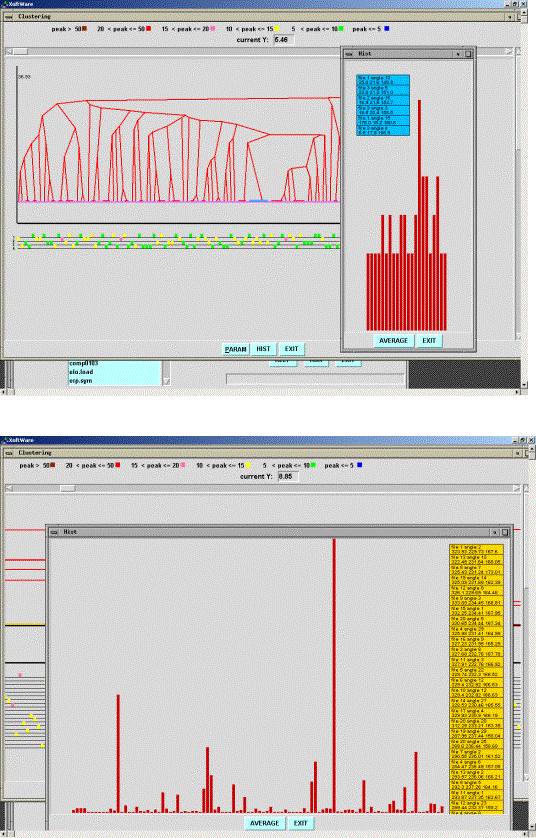
interatomic distance (Fig. 1). A selection of a cluster in the histogram
indicates it in the cluster tree, gives the corresponding angle values and can
provide with the atomic models rotated respectively.
First tests
This procedure has been tested first with a synthetic case and then was
successfully applied to several experimental cases where the structure could
not be solved previously by conventional molecular replacement procedures.
In this first series of tests, a simple but usual situation was
simulated when the model is quite poor to give a strong signal in the rotation
function. The N-terminal end (first 100 residues from 689 in the complete
model) of a large protein, the elongation factor G (Aeverson et al.,
1994) was used as the search model. Corresponding crystals have the symmetry P212121, unit cell parameters a =
75.6, b = 106.0, c = 116.6 Ĺ. The rotation function was calculated for the same
model but in different resolution ranges : 4 – 15 Ĺ, 4 – 10 Ĺ, 4 – 8 Ĺ, 5 – 10
Ĺ. While individual rotation functions do not allow to identify the solution
(Table 1), the merging of the rotation peaks in the cluster tree and cluster
selection with the distance of 5 degrees, showed the correct orientation
unambiguously (Fig. 1). This peak is stable in a large range of the distance
cut-off. It can be noted also that, being presented as they are in the rotation
function files, not all angles of this cluster are close between themselves
from the first look (Table 1). When the distance cut-off decreases to 3 degrees the cluster is
reduced to three closest peaks (first three lines of the Table 1) very close to
the exact answer.
The
second series of tests was done with experimental data of ER-1 protein
(Anderson et al., 1996) called by the authors “A challenging case for
protein crystal structure determination”. This small 40 amino-acids protein
crystallises very densely in the space group C2 with the unit cell parameters a
= 53.91, b = 23.08, c = 23.11 Ĺ, b = 110.4°. The authors failed to identify the correct rotation using
available 20 NMR models.
In this case of a small protein the
data of the resolution of at least 8Ĺ and lower should be excluded from the
calculation due to a very strong influence of the bulk solvent on structure
factors; Anderson et al. found that the best resolution cut-off is even
7 Ĺ. Two sets of rotation functions were calculated varying the model, one at
the resolution of 3-8 Ĺ, and the second at the resolution of 4-8 Ĺ. Similarly
to the previous report (Anderson et al., 1996), AMoRe did not find the
solution in any of these runs. In fact, the lists of the rotation peaks contain
orientations close to the correct one; translation functions calculated with
them also contain the correct position; however, it is not possible to
recognise the answer among many tens of variants with a better correlation,
sometimes even essentially better.
Multiple rotation function analysis with the functions calculated at 3-8
Ĺ shows an extremely strong peak when the angular distance is equal to 9
degrees (Fig. 2, peak contribution to the cluster size was weighted by their
height). When the angular distance is decreased to about 5 degrees, the cluster
is split into 2 subclusters where the larger one is closer to the correct
solution. If the orientation of the first model is chosen from this cluster,
the translation function and the intermolecular distance allow immediately to
identify the solution (Table 2) even by traditional translation search.
For the rotation functions
calculated at the resolution 4-8 Ĺ, the peaks are weaker and further from the
correct orientation and their cluster analysis shows the answer unambiguously
only at a quite high interangular distance, of order of 10°. A common analysis
of all 40 rotation functions (20 at every resolution shell, 4-8 Ĺ and 3-8 Ĺ)
showed again the correct orientation clearly.
In general, from our experience is
seems to be efficient to start the clustering analysis from relatively high
interangular distances, of about 10 degrees, to find the principal cluster or
clusters and then decrease the distance level to select the solution (or few
possible solutions, in general case) inside them. Very high interangular
distance, of 20 degrees and higher, starts to put together the peaks which have
nothing in common and can lead to misleading results.
>The third series of tests has been
done with experimental data of thioredoxin h from Chlamydomonas reinhardtii (A. Aubry, personal communication) where it was not possible to solve
the structure by the conventional molecular replacement using available 23 NMR
models (the structure has been solved in a different way, the paper is in
preparation). In this case, when the standard AMoRe protocol does not give the
answer, the clustering with the distance level of 3° and higher shows
immediately the correct orientation corresponding to the cluster of the size 3
times larger that the size of the next cluster. Use of an existing
noncrystallographic symmetry doubled the signal. Details of this test and some
others will be discussed elsewhere.
Conclusions
Cluster analysis of multiple rotation functions can be useful in many
practical situations when searching for the model orientation with imperfect
models. A relatively random distribution of noisy peaks allows to identify the
signal which appears systematically (but, maybe, weakly) in the rotation
functions. Naturally, the cluster analysis gives an information which is
definitely more reach than a single orientation for such or such model. The use
of this information for further steps of molecular replacement, specially for
the translation function, will be discussed elsewhere.
Acknowledgment
The authors thank C. Lecomte for his
interest to the project, A. Aubry for the thioredoxin data available before
their publication, and L. Torlay for the technical help.
References
Ćvarsson, A., Braznihnikov, E., Garber, M.,
Zhelnotsova, J., Chirgadze, Yu., Al-Karadaghi, S., Svensson, L.A. & Liljas,
A.(1994). EMBO Journal, 13,
3669-3677.
Anderson, D.H., Weiss, M.S. &
Eisenberg, D. (1996) Acta Cryst., D52, 469-480.
Brünger, A.T. (1990) Acta Cryst., A46, 46-57.
Chang, G. & Lewis,
M. (1997) Acta Cryst.,
D53, 279-289.
DeLano, W.L. & Brünger, A. (1995). Acta Cryst., D51, 740-748.
Kissinger, C.R., Gehlhaar, D.K. & Fogel, D.B.
(1999) Acta
Cryst., D55,
484-491.
Lunin, V.Y. &
Urzhumtsev, A.G. (1999). CCP4 Newsletter
on Protein Crystallography, 37,
14-28.
Navaza, J. (1994) Acta Cryst.,
A50, 157-163.
Navaza, J. &
Vernoslova, E. (1995) Acta
Cryst., A51,
445-449.
Ousterhout, J.K. (1993) "Tcl
and the Tk Toolkit". Addison-Wesley Publishing Company.
Rossmann, M.G. (1972) The Molecular Replacement Method., Gordon
& Breach; New York, London, Paris.
Rossmann, M.G. (1990) The Molecular Replacement Method. Acta Cryst., A46, 73-82.
Urzhumtseva, L.M., Urzhumtsev, A.G. (1997) J.Appl. Cryst., 30,
402-410.
Table 1. Rotation
functions analysis for the N-terminal end of the EFG. The correct solution is
(27.6, 21.9, 148.3).
|
Resolut. limits |
Sequen. N of the peak |
a,b,g |
Height of the peak |
Height of the1st peak |
Height of the 2nd peak |
|
4-10 |
10 |
25.8,
21.6, 148.9 |
10.0 |
13.2 |
12.4 |
|
5-10 |
5 |
23.0,
21.2, 151.0 |
11.3 |
14.1 |
13.1 |
|
4-15 |
16 |
18.9,
21.6, 153.7 |
13.4 |
18.5 |
15.7 |
|
5-10 |
3 |
18.5,
20.4, 158.5 |
11.3 |
14.1 |
13.1 |
|
4-10 |
15 |
176.0, 18.2,
180.8 |
9.8 |
13.2 |
12.4 |
|
5-10 |
4 |
6.8,
17.9, 166.9 |
11.3 |
14.1 |
13.1 |
Table 2. Translation
search for the ER1 (first NMR model)
for the rotation angles defined by the multiple function analysis as
(116.2, 73.3, 209.9). The correct orientation found from the optimal model
superposition is (113.3, 77.2, 200.3) and the position is (0.3151, 0.0,
0.4892). Appropriate solutions are indicated by *.
|
Peak N |
a,b,g |
Molecular position |
Correlat. |
Intermolec. distance |
|
1 |
113.1 77.9
202.0 |
0.4260 0.0 0.4493 |
49.2 |
7.0 |
|
2** |
110.8 74.6
207.7 |
0.3209 0.0 0.4936 |
37.5 |
14.6 |
|
3* |
114.2 76.6
203.6 |
0.3823 0.0 0.4902 |
35.4 |
12.9 |
|
4 |
113.7 77.9
204.8 |
0.4714 0.0 0.3263 |
30.7 |
7.1 |
|
5 |
112.8 77.7
207.6 |
0.0837 0.0 0.3635 |
27.7 |
13.2 |
|
6 |
113.0 72.9
210.2 |
0.2043 0.0 0.4097 |
26.8 |
12.4 |
Fig. 1. Copy of the
screen during the program session when comparing several rotation functions for
the EFG N-terminal model (see Section ‘First Tests’). The correct orientation
corresponds to the cluster (shown in light bleu in the cluster tree) with the
largest cluster size (shown in the inserted window). Initial rotation angles
(as they are done in the or1.s files) are shown in bleu frame. Several
parallel lines with squares below the cluster tree show the rotation peaks in
different rotation functions with their height indicated by colour. A variable
cut-off interangular distance is indicated by a pink line above the zero level
(black line)
Fig. 2. Cluster size
analysis for the ER-1 protein (see Section ‘First Tests’). The correct
orientation corresponds to the cluster with the largest cluster size. Note the
contrast of the signal. Final rotation angles (corresponding to sequential
rotation defined in tabl1.s and or1.s files) are shown in yellow
frame.
 |
|
Newsletter contents... |