MASC:
A Combination of Multiple-Wavelength Anomalous Diffraction
& Contrast Variation
W. Shepard†, M. Ramin†, R. Kahn§, & R. Fourme
†
LURE, B.209D, Université Paris Sud, 91405 Orsay, France.§
Institut de Biologie Structurale, 41 Avenue des Martyrs, 38027 Grenoble, France.
1. Introduction
Contrast variation methods have primarily been applied and developed in low angle scattering studies as a means of extracting information on the shape of a particle dispersed in a solvent medium (for a review see Williams et al. , 1994). This method deals with the changes invoked in the scattered intensities of a small angle scattering experiment when the density of the particle is varied relative to its solvent medium. The difference between the particle and solvent densities is defined as the "contrast" (Stuhrmann & Kirste, 1965; Ibel & Stuhrmann, 1975). The term "density" in this context refers to the electronic density in an X-ray scattering experiment, the isotopic substitution ratio (H/D) in a neutron scattering exp eriment, or any other physical density which scatters the incident beam.
Contrast variation techniques can be extended to macromolecular crystal systems since such crystals typically consist of 30-70% solvent, which is a phase of rapidly interchanging molecules. Bragg & Perutz (1952) applied such met hods to a haemoglobin crystal and observed changes in the intensities of low resolution X-ray reflections after altering the electronic density of the mother liquor. In particular, they related these changes to the Fourier transform of the solvent accessi ble regions of the crystal. In other words, the data from a contrast variation series provides information on the macromolecular envelope.
Others have since applied contrast variation techniques in either X-ray or neutron distraction experiments to glean low resolution structures from macromolecular crystals (Harrison, 1969; Jack, Harrison & Crowther, 1975; Moras e t al., 1983; Roth et al., 1984: Bentley et al., 1984; Podjarny et al., 1987). In particular, Carter et al. (1990) used a formalism which separated the diffraction effects of the molecular envelope and the internal fluctuations (Bricogne, unpublished) in t he direct phase determination of the molecular envelope of tryptophanyl-tRNA synthetase.
Anomalous dispersion has also been employed in small angle scattering experiments to produce contrast variation. Examples on biological systems are the harnessing of the iron K-edge in ferritin (Stuhrman, 1980) and the phosphorous K -edge in ribosomes (Hütsch, 1993). In crystallography, the use of anomalous scattering effects from the solvent has been suggested by Wyckoff and others where it could be used as a supplement to a standard contrast variation series (Dumas, 1988; Crum ley, 1989; and Carter et al., 1990). However, in these cases, the anomalous scattering was still restricted at a single wavelength. The possibility of exploiting the
full potential of anomalous scattering at several wavelengths was originally put forward by Bricogne (1993).
2. Theoretical principles of contrast variation
![]() Here only an outline of the theoretical principles will be given. Readers wishing for a fuller account are referred to Fourme et al. (1995). The starting point
of what we call MASC (Multiple- wavelength Anomalous Solvent Contrast) is the basic principles of contrast variation where the macromolecular crystal lattice is assumed to be biphasic: one region of the unit cell is occupied by the macromolecule domain U
(Figure 1a), and the other domain V-U (Figure 1b) is occupied by the solvent which is in a liquid-like state of rapid exchange. The domain containing the macromolecule is presumed to be ordered, whereas the solvent regions are presumed to be completely d
isordered.
Here only an outline of the theoretical principles will be given. Readers wishing for a fuller account are referred to Fourme et al. (1995). The starting point
of what we call MASC (Multiple- wavelength Anomalous Solvent Contrast) is the basic principles of contrast variation where the macromolecular crystal lattice is assumed to be biphasic: one region of the unit cell is occupied by the macromolecule domain U
(Figure 1a), and the other domain V-U (Figure 1b) is occupied by the solvent which is in a liquid-like state of rapid exchange. The domain containing the macromolecule is presumed to be ordered, whereas the solvent regions are presumed to be completely d
isordered.
We define ![]() as the electronic density of the solvent volume, which is constant since this region is flat and featureless.
as the electronic density of the solvent volume, which is constant since this region is flat and featureless. ![]() is the Fourier transform
of the indicator function
is the Fourier transform
of the indicator function ![]() defined as equal to 1 inside the volume U and 0 elsewhere (Bricogne, 1974). It should be noted that
defined as equal to 1 inside the volume U and 0 elsewhere (Bricogne, 1974). It should be noted that ![]() = GV-U(h) when
= GV-U(h) when ![]() , such that GV-U(h) is the Fourier transform of the complementary indicator function
, such that GV-U(h) is the Fourier transform of the complementary indicator function ![]() which corresponds to the region occupied by the solvent. The total structure factor, F(h), can be written as the sum of two components: one from the ordered regions of the crystal,
which corresponds to the region occupied by the solvent. The total structure factor, F(h), can be written as the sum of two components: one from the ordered regions of the crystal, ![]() , an
d the other from the solvent.
, an
d the other from the solvent. ![]() These two components are related since the volume occupied by either the macromolecule or the solvent are by definition mutually exclusive.
These two components are related since the volume occupied by either the macromolecule or the solvent are by definition mutually exclusive.
![]()
Fp(h) is also the Fourier transform of the macromolecule in a vacuum (Figure 1c), and it can be expressed as the sum of the ·rpÒGU(h) and D(h), the latter which is the Fourier tra
nsform of the internal density fluctuations from the mean density inside the domain U (i.e. ![]() see figure 1e).
see figure 1e).
![]()
Substituting in this expression for ![]() gives
gives
![]()
The term ![]() is defined as the contrast (Stuhrmann & Kirste, 1965), and when it is equal to zero the system is said to be at the constrast matching point (see figure 1d) whereby only the intern
al electronic density fluctuations contribute to the overall structure factor. A demonstration of this expression can be found in Carter et al. ( 1990).
is defined as the contrast (Stuhrmann & Kirste, 1965), and when it is equal to zero the system is said to be at the constrast matching point (see figure 1d) whereby only the intern
al electronic density fluctuations contribute to the overall structure factor. A demonstration of this expression can be found in Carter et al. ( 1990).
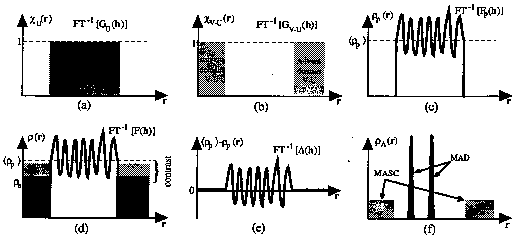
Figure 1.
The 1-dimensional slices of different components in contrast variation theory: a) Indicator function of the ordered domain, U, containing the protein. b) Indicator function of the disordered domain, V -U, containing the solvent. c) The electronic density of only the ordered domain, U. This corresponds to the macromolecule in a vacuum. d) The electronic density for both the macromolecule and solvent regions. Three different electronic densities of the s olvent are represented by the three shades of grey. The contrast is shown for one of these. e) The internal electronic density fluctuations inside the macromolecule. f) The anomalous electronic density for both the MAD and MASC cases.
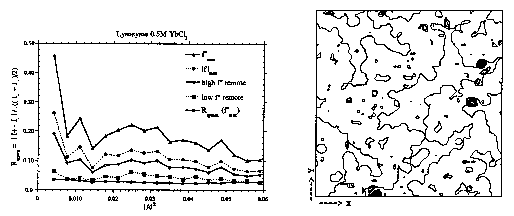
Figure 2.
On the left, anomalous R-factors for MASC data of HEW lysozyme in 0.5M YbCl3. Four wavelengths at the Yb LII I-edge plus the R-factor for true symmetry related reflections (i.e. respecting the differences between I+ and I-). On the right, ordered sites of Yb3+ ions in HEW lysozy me crystals. A phased anomalous Fourier map is superimposed on to a map of the protein envelope. Dark spots show Yb3+ positions in crevices and near the surface of the protein.
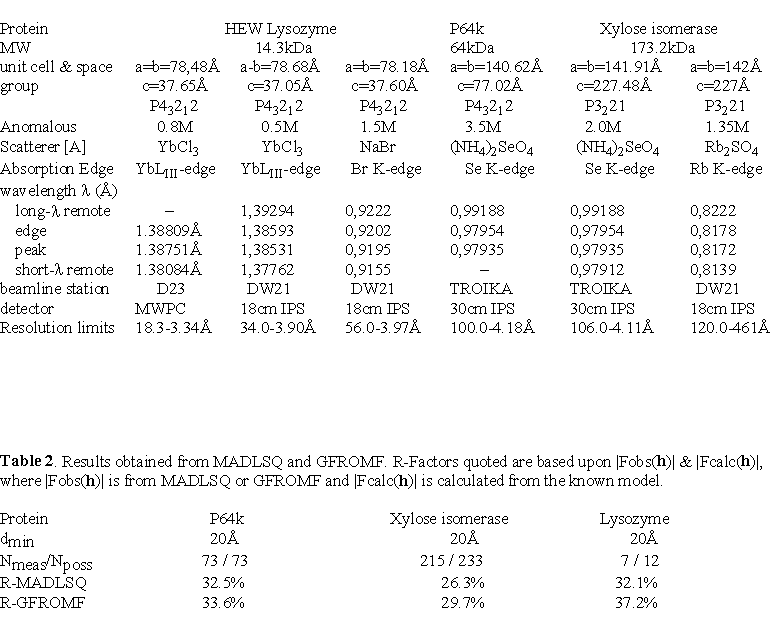
Table 1.
Extension to the case where anomalous scatterers are present in the solvent can be done using the seminal idea of Karle (1980) where the structure factors are separated into wavelength independent (f°) and depende nt parts
![]()
![]()
For our discussion here, we assume that there is only one anomalous scatterer which is randomly dispersed in the solvent domain V-U such that it has a uniform distribution and no ordered sites bound to the surface of the macromolecule. For simplicity, we also assume that any scattering factor at low resolution is constant with respect to scattering angle to within a first order approximation. The density of the anomalous scatterers in the solvent can be treated as a complex quantity, lrsA, which is dependent upon wavelength,
![]()
and where ![]() is the normal electronic density of the anomalous scatterer. The total electonic density of the solvent,
is the normal electronic density of the anomalous scatterer. The total electonic density of the solvent, ![]() , becomes a function of the wavele
ngth, and can be separated into wavelength independent and dependent parts,
, becomes a function of the wavele
ngth, and can be separated into wavelength independent and dependent parts,
![]()
Note that the term
0rs includes the normal scattering part of the anomalous scatterer. Thus one obtains,![]()
The terms in between the first set of brackets represent the wavelength independent part of the overall structure factor, denoted °F(h). It includes the envelope, constrast and fluctuation terms. The second set of brackets is wav
elength dependent, and incorporates the envelope and the anomalous structure factors of A, ![]() Note that the wavelength dependent contribution is substracted from the normal scattering part indicating that the anomal
ous and dispersive structure factors of A are applied to the Fourier transform of the indicator function of the solvent accessible domain, - G
Note that the wavelength dependent contribution is substracted from the normal scattering part indicating that the anomal
ous and dispersive structure factors of A are applied to the Fourier transform of the indicator function of the solvent accessible domain, - G
By defining ![]() one generates a expression of the overall structure factor similar to the starting point used for the algebraic MAD method (Hendrickson, 1985) where
one generates a expression of the overall structure factor similar to the starting point used for the algebraic MAD method (Hendrickson, 1985) where
![]()
The substitution of ![]() for °F
for °F
![]()
where, ![]()
A MASC experiment has an advantage over other contrast variation methods, since the contrast variation is generated by inducing a physical change. This eliminates the possibility of changes in the crystal lattice due to varying ioni c strength, pH, precipitant concentration, etc... which can arise in a chemical contrast series, and thus enforces strict isomorphism.
3. Strength of the anomalous signal in MASC
The strength of the signal in an anomalous contrast variation series can be quantified in a similar way to those in the MAD method, i.e. by measuring differences between Bijvoet pairs (anomalous or
lf" contribution) and wavelengths (dispersive or lf' contribution). Intuitively, the magnitude of the anomalous signal in a MASC experiment is expected to vary consi derably with resolution, being very large in the lowest resolution shells and then diminishing rapidly with increasing resolution. One also expects the anomalous signal to be directly proportional to the concentration of the anomalous scatterer in the sol vent accessible volume. Furthermore, the signal will be maximised at the point of contrast matching. By making a certain number of approximations, it is possible to derive expressions for and calculate the expected anomalous and dispersive ratios (Fourme et al., 1995), but for the purpose of succintness only the final expressions will be given here. Thus for anomalous and dispersive differences one gets†,
![]()
and
![]()
Clearly, the anomalous signal is dependent on a number of factors, such as the molar concentration of the anomalous scatterers, [A] and the magnitudes of f" and
Df'. However, the resolution, s, has the strongest effect on the anomalous signal which drops away as a function of 1/s2 and exp(-Bs2/4). The term exp(-Bs2/4) represents a Gaussian smoothing of the
Where:
![]()
envelope boundary, where B is pseudo-temperature factor which defines the thickness of the interface rather than the temperature factor of the macromolecule or solvent. The signals are also somewhat dependent upon the molecular weight, but a 100kDa protein will still produce 68% of the signal of a 10kDa protein. For a hypothetical case of a 50kDa protein in 3.5M (NH
4)2Se O4, where f"=7.0e-,
4. Experimental
As a MASC experiment utilises the variation of f' and f", data collection should ideally be carried out at X-ray wavelengths near absorption edges of the anomalous scatterer. Thus the requirements are very similar to a MAD exper
iment - i.e. tuneable X-rays with a narrow band pass (![]() 10
10
A variety of anomalous scatterers may be used in a MASC experiment, and the most suitable ones will depend on the crystallisation conditions of the macromolecule. Analogues of the precipitating agent are good choices since such comp ounds are less likely to perturb the crystalline lattice (e.g. selenate for sulphate, bromide for chloride, tribromoacetate for acetate etc...). To date, MASC data have been collected on crystals of three proteins of differing molecular weights and with a variety of different anomalous scatterers (see Table 1). In order to develop the MASC method, all of the cases are known crystal structures, which allows the experimental results to be compared with the correct envelope transform moduli and phases. In ea ch of the experiments described below, the X-ray diffraction data were recorded at the wavelengths corresponding to the |f'|
max and the f"max which were determined from the X-ray fluorescence spectra from a solution of the anomalous scatterer, as well as for at least one wavelength remote of the absorption edge. A small beamstop (≈2-3mm) was mounted and aligned just in front of the e ntrance window of the detector. Where possible, the crystallographic axes were aligned so that Bijvoet pairs could be measured on the same image. Below we describe in detail the experiments and the results for only two anomalous scatterers.4.1 Hen egg white lysozyme co-crystallised in YbCl
3The very first MASC experiment was conducted on single crystals of lysozyme directly crystallised from solutions of 0.3-0.5M YbCl
3. This combin ation was chosen because of the
Figure 3.
Fluorescence and absorption effects of the diffraction pattern from a xylose isomerase crystal in 2M (NH4)2< /FONT>SeO4 recorded at four different wavelengths about the Se K-edge and of the same region of reciprocal space.
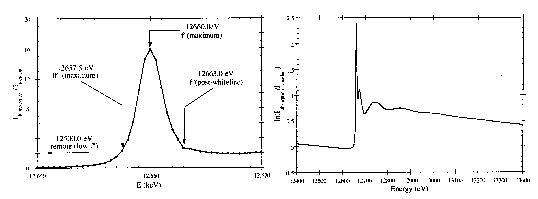
Figure 4.
Whiteline structure of the Se K-edge of 0.1M (NH4)2SeO4 recorded on the TROÏKA beamline station, ESRF, France (left), and the extended absorption spectra recorded at LURE, Orsay, France (right).
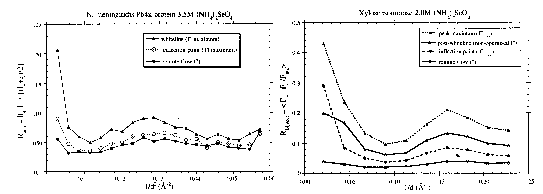
Figure 5.
Anomalous it-factors as a function of resolution for P64k and xylose isomerase in (NH4)2SeO4. Please note that the resolution is broken down as a function of l/d2 for P64k and l/d for xylose isomerase.
ease of obtaining crystals and their robustness, as well as for the white line structure of the Yb L
III-edge. The Yb3+ ion concentration could be increased to 0.8M using vapour diffusion techniques before the crystal quality would deteriorate. X-ray diffraction data were collected at three wavelengths including one remote on the high energy side of' the Yb LIII-edge on the D23 station (Kahn et al., 1986) at LURE-DCI (Orsay, France). Bijvoet ratios are shown in Figure 2. The results confirmed the large anomalous signal at low resolution as expected by theory. At t he wavelength corresponding to the maximum of f", the Bijvoet ratio reaches ≈50% for the lowest resolution shell and then diminishes sharply with increasing resolution. The internal agreement between true equivalent reflections is within ≈1-3% , implying that the anomalous signal is real, reproduceable and not artifact of either the data processing or the beamstop shadow. The anomalous signal however extends well beyond 10Å resolution indicating that some Yb3+ ions have bound to the protein. Anomalous difference Patterson maps did not reveal the positions of three bound Yb3+ ions, which were eventually found in a phased an omalous difference Fourier map (Fig. 2). The reason for this might be because the diffraction data is only4.2 P64k and xylose isomerase in (NH
4)2SeO4P64k is a 64kDa outer membrane protein from Neisseria meningitidis currently under study in our lab (Li de la Sierra et al., 1994; Li de la Sierra et al., 1997), and it crystallises from ammonium sul phate solutions. Xylose isomerase also crystallises from ammonium sulphate solutions but as a tetramer (173.2kDa) in the asymmetric unit (Rey et al., 1988). Both of these proteins represent large macromolecular structures on the scale of those typically s olved by the MAD method. Ammonium sulphate in the mother liquor of the crystals could be substituted with multimolar concentrations of ammonium selenate via simple soaking techniques. Crystals of both proteins could withstand 3.5M (NH
4)2SeO4, which brings the solvent electronic density equal to the average protein electronic densit y, i.e. the contrast matching point. This allowed us to collect MASC data at the Se K-edge of selenate which features a white-line structure at a wavelength near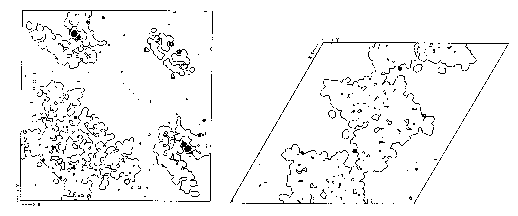
Figure 6.
Ordered sites of selenate ions in P64k (left) and XI (right). In each case a phased anomalous Fourier map is superimposed on to a map of the protein envelope. Dark spots on each map show selenate posi tions in crevices and near the surface of the protein.
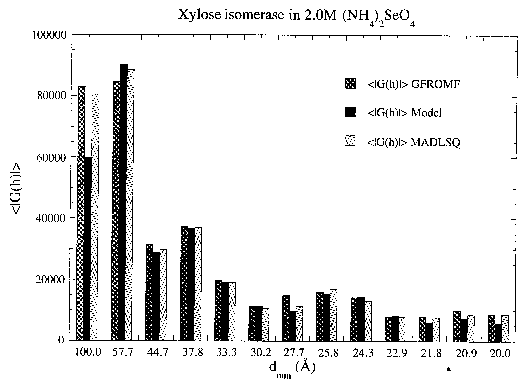
Figure 7.
Agreement as a function of resolution of |GU(h)| values of xylose isomerase in 2.0M (NH4)2SeO4 calculated from MADLSQ, GFROMF and its model.
minimise the absorption and fluorescence effects, a number of precautions were taken: i) Crystals were rapidly rinsed or washed in an analogous sulphate solution just prior to freezing, thus removing mother liquor containing any excess
selenate surrounding the crystal, ii) smaller crystals were used to reduce the amount of absorption relative to the crystal volume, and iii) finer oscillation angles were recorded for the wavelength corresponding to the maximum of f" to improve the signal
to background ratio. There are other tactics which could be employed to minimise fluorescence effects. For X-ray energies used in typical crystallography experiments (0.5Å - 2.0Å), the fluorescence yield after absorbing an incident photon is
2-3 times higher for the K-edges than for the L-edges (Kortright, 1986). For example, the fluorescence yield is ![]() 60%, for Se at its K-edge, whereas the fluorescence yield is only
60%, for Se at its K-edge, whereas the fluorescence yield is only ![]() 20% for Yb at one of its L-edges. Another method to reduce fluorescence effects is simply to increase the sample-to-detector (D) distance since fluorescence which is radiated isotropically will fall off as l/D
20% for Yb at one of its L-edges. Another method to reduce fluorescence effects is simply to increase the sample-to-detector (D) distance since fluorescence which is radiated isotropically will fall off as l/D
The anomalous signal for both proteins follows the expected trend, being very large for the Bijvoet pairs at lowest resolution and decreasing rapidly with increasing resolution (see Figure 5 ). At higher resolution, the Bijvoet rati os for both proteins are of the order of the internal agreement, but despite this low anomalous signal, up to 12 possible selenate ion sites have been located from phased anomalous difference Fourier maps in the P64k crystals (see Figure 6). Similarly, se veral selenate ion sites have been found in the crystals of xylose isomerase. All sites are at or near the macromolecular boundary, often in crevices, and their relative occupancies vary considerably. The existence of ordered anomalous sites appears to be more general than expected, but it opens up a potential of phasing to higher resolution once a model for envelope is determined.
5. Extracting, |G
U(h)| from MASC dataTwo methods have been utilised to extract the moduli of G
U(h) from multiple-wavelength diffraction data. One uses the algebraic equations in the MAD method as imp lemented in the program MADLSQ (Hendrickson, 1985), and the other uses the program GFROMF (Carter & Bricogne, 1987), which is designed to extract the Prior to using either method of extracting the ![]() , the X-ray data were set on a common scale using the program SCALA (Evans, 1993). The data were scaled in two steps: i) an internal scaling f
or each wavelength to correct for incident beam fluctuations and sample decay, and ii) a pseudo-local scaling between a reference wavelength (low f") and the other wavelengths to minimise absorption effects.
, the X-ray data were set on a common scale using the program SCALA (Evans, 1993). The data were scaled in two steps: i) an internal scaling f
or each wavelength to correct for incident beam fluctuations and sample decay, and ii) a pseudo-local scaling between a reference wavelength (low f") and the other wavelengths to minimise absorption effects.
5.1 MADLSQ
As mentioned above, the program MADLSQ, which was originally designed for multiple- wavelength diffraction data, solves the set of equations by non-lineal least-squares for ![]() and the phas
e difference
and the phas
e difference ![]() . For MASC data,
. For MASC data, ![]() replaces
replaces ![]() , and the phase difference becomes
, and the phase difference becomes ![]() . The pr
ogram also has the ability to refine or fix the
. The pr
ogram also has the ability to refine or fix the
values of f' and f" of the different wavelengths. Results are shown in Figure 7 for the data collected on xylose isomerase crystals soaked in (NH
4)2SeO4 and as compared to the5.2 GFROMF
In chemical contrast variation studies, the program GFROMF (Carter & Bricogne, 1987) extracts the ![]() from the diffraction data
from the diffraction data ![]() for i=l,...,N
where i corresponds to a different solvent density,
for i=l,...,N
where i corresponds to a different solvent density, ![]() . To extend this to multiple wavelength cases, we simply substitute in for the contrast series
. To extend this to multiple wavelength cases, we simply substitute in for the contrast series ![]() where
where
|![]()
The GFROMF scheme carries out the non-linear least-squares refinement of ![]() , X(h) and Y(h) from scaled data summed over all contrasts, and minimises the function,
, X(h) and Y(h) from scaled data summed over all contrasts, and minimises the function,
![]()
where
sobs(h) is standard deviation ofThe original program was modified to incorporate anomalous scattering contributions such that,
![]()
Tests executed on simulated MASC data of kallikreen (52kDa), at three different contrasts of selenate and three wavelengths per contrast, returned exact values of ![]() , X(h) and Y(h)
of the simulated observed data. With experimental data, the results gave R-factors of ≈30-35% for P64k and xylose isomerase crystals (see Table 2). This level of agreement is satisfactory considering that many of the parameters are unrefined. In par
ticular. the values of
, X(h) and Y(h)
of the simulated observed data. With experimental data, the results gave R-factors of ≈30-35% for P64k and xylose isomerase crystals (see Table 2). This level of agreement is satisfactory considering that many of the parameters are unrefined. In par
ticular. the values of ![]() employed were derived from previous runs of MADLSQ, and theoretical values of the contrast were used rather than allowing them to refine. The scale factors between different wavelengths
employed were derived from previous runs of MADLSQ, and theoretical values of the contrast were used rather than allowing them to refine. The scale factors between different wavelengths
![]() were set to unity since the data were already set on a common scale. In principle, all of these parameters should be refineable in the GFROMF scheme, even though the number of observations in t
he lower resolution shells is not overly large. What is certain is that prior precise knowledge of the values of the contrasts,
were set to unity since the data were already set on a common scale. In principle, all of these parameters should be refineable in the GFROMF scheme, even though the number of observations in t
he lower resolution shells is not overly large. What is certain is that prior precise knowledge of the values of the contrasts, ![]() is important to extract
is important to extract ![]() values of satisfa
ctory quality.
values of satisfa
ctory quality.
6. Phasing G-moduli
Previous methods of phasing ![]() from either X-ray contrast variation series (e.g. Carter, et al ) or H/D substitution contrast variation series (Moras et al., 1983; Roth et al., 1984; Bentl
ey et al., 1984; Podjarny et al., 1987; Roth, 1991) employed the assumptions that the set of
from either X-ray contrast variation series (e.g. Carter, et al ) or H/D substitution contrast variation series (Moras et al., 1983; Roth et al., 1984; Bentl
ey et al., 1984; Podjarny et al., 1987; Roth, 1991) employed the assumptions that the set of ![]() behave much in the same way as the structure factors of small molecule crystal structures. Hence such attempts have use
d the programs of traditional direct methods of small molecule crystallography. As a starting point, we have also examined this strategy in preliminary trials for phasing a set of
behave much in the same way as the structure factors of small molecule crystal structures. Hence such attempts have use
d the programs of traditional direct methods of small molecule crystallography. As a starting point, we have also examined this strategy in preliminary trials for phasing a set of ![]() from MASC data, but it is clear
that the limited success with these methods necessitates a re-examination of the phasing methods used up to now.
from MASC data, but it is clear
that the limited success with these methods necessitates a re-examination of the phasing methods used up to now.
Using 1664 calculated ![]() up to 10Å resolution from a model of xylose isomerase, phase sets for the |G
up to 10Å resolution from a model of xylose isomerase, phase sets for the |G
The limited success obtained from the use of traditional direct methods is not surprising considering that such methods are based on a variety of assumptions which are not valid for a set of ![]() . For example, envelopes are not point scatterers, as can be assumed for atoms. Also an envelope also does not represent a random distribution of scatterers; quite the contrary, by definition of the biphasic model, the scatterers are confined inside the
volume of the solvent. Consequently, a set of
. For example, envelopes are not point scatterers, as can be assumed for atoms. Also an envelope also does not represent a random distribution of scatterers; quite the contrary, by definition of the biphasic model, the scatterers are confined inside the
volume of the solvent. Consequently, a set of ![]() does not follow Wilson statistics. In addition, normalisation of'
does not follow Wilson statistics. In addition, normalisation of' ![]() can not be accomplished as in traditional methods becaus
e of the relatively few reflections at low resolution and their very large dynamic range. Despite these differences with small molecules, a set of
can not be accomplished as in traditional methods becaus
e of the relatively few reflections at low resolution and their very large dynamic range. Despite these differences with small molecules, a set of ![]() has the advantage in being complete with relatively few reflectio
ns (i.e. there are only a total of 73 unique reflections to 20Å resolution for P64k).
has the advantage in being complete with relatively few reflectio
ns (i.e. there are only a total of 73 unique reflections to 20Å resolution for P64k).
The problem of phasing a set of ![]() clearly needs to be readdressed. We are currently considering other methods towards phasing
clearly needs to be readdressed. We are currently considering other methods towards phasing ![]() , and the use of Maxim
um Entropy and Likelihood ranking to test envelope hypotheses. The literature shows an increasing interest in the field of low resolution phasing. Some of these methods approximate globular proteins as spheres or a few large Gaussian spheres (Andersson &a
mp; Hovmöller, 1996); Harris, 1995; Lunin et al., 1995; Urzhumtsev et al., 1996), or as a gas of hard sphere point scatterers (Subbiah, 1991); Subbiah, 1993).
, and the use of Maxim
um Entropy and Likelihood ranking to test envelope hypotheses. The literature shows an increasing interest in the field of low resolution phasing. Some of these methods approximate globular proteins as spheres or a few large Gaussian spheres (Andersson &a
mp; Hovmöller, 1996); Harris, 1995; Lunin et al., 1995; Urzhumtsev et al., 1996), or as a gas of hard sphere point scatterers (Subbiah, 1991); Subbiah, 1993).
7. Conclusions & Perspectives
It has been demonstrated that contrast variation in macromolecular crystallography can be generated using anomalous dispersion techniques in a MAD-like experiment. The method benefits from the strict isomorphism imposed by the e xternal physical chance of the wavelength of the X- rays applied to a single sample. This is clearly advantageous over a chemical contrast series experiment which typically requires several samples soaked in different media. and which risks destroying any isomorphism.
From the studies presented here, large anomalous signals are observed in the lowest resolution shells. In all of the cases studied to date, the anomalous signal extends to higher resolution indicating the presence of ordered anomalo
us scattering sites. Such sites have little effect at low resolution, and they are a bonus in a MASC experiment because they may provide a path for phasing the 3D structure to higher resolution once the envelope is known. Extracting the set of ![]() from MASC data can be accomplished using two different procedures; one based on the algebraic equations of multiple-wavelength diffraction data (MADLSQ) and the other based on the equations derived from a chemical contrast
variation series (GFROMF).
from MASC data can be accomplished using two different procedures; one based on the algebraic equations of multiple-wavelength diffraction data (MADLSQ) and the other based on the equations derived from a chemical contrast
variation series (GFROMF).
The process of phasing a set of ![]() needs further attention. Traditional direct methods. which are intended for small molecule structures, are not suitable for this type of phase problem. If th
e phasing step of a set of
needs further attention. Traditional direct methods. which are intended for small molecule structures, are not suitable for this type of phase problem. If th
e phasing step of a set of ![]() can be dealt with, then the combination of anomalous dispersion and contrast variation techniques can lead to a general method for low resolution phasing of very large macromolecules in
cluding those beyond the scope of MIR and MAD methods. Finally, knowledge of the macromolecular envelope will help phase the structure to higher resolution.
can be dealt with, then the combination of anomalous dispersion and contrast variation techniques can lead to a general method for low resolution phasing of very large macromolecules in
cluding those beyond the scope of MIR and MAD methods. Finally, knowledge of the macromolecular envelope will help phase the structure to higher resolution.
Acknowledgements
We thank A. Thompson, A. Gonzalez, G. Grübel, D. Abernathy & M. Lehmann for support during the experiments on the TROÏKA beamline at the ESRF, Grenoble, France. We are also grateful to D. Ragonnet. D. Chandesris and th e SEXAFS group at LURE for assistance with the DW21 beamline.
References
Andersson, K.M. & Hovmöller, S. (1996) Acta Cryst. D52, 1174-1180.
Bentley, G.A., Lewit-Bentley, A., Finch, J.T., Podjarny, A.D. & Roth, M. (1984). J. .Mol. Biol. 176, 55-75.
Bragg, W.L. & Perutz, M.F. (1952) Acta Cryst. 5, 277- 289.
Bricogne, G. (1974) Acta Cryst. A30, 395-405.
Bricogne, G. (1993) Acta Cryst. D49, 37-60.
Carter, C.W., Jr. & Bricogne, G. (1987) GFROMF: A computer program for scaling and estimating envelope structure factors from contrast variation data. Dept. of Biochemistry, CB#7260, University of North Carolina at Chapel Hill, Chap el Hill, NC 27599-7260, USA
Carter, C.W., Crumley, K.V., Coleman, D.E., Hage, F. & Bricogne, G. (1990) Acta Cryst. A46, 57-68.
Crumley, K.V. (1989) M. Sc. Thesis, University of North Carolina, Chapel Hill. USA.
Dumas, C. (1988) Thèse de Doctorat des Sciences Naturelles, Université Paris-Sud, Centre Scientifique d'Orsay, France.
Evans, P. R. (1993) In Proceedings of the CCP4 Study Weekend on Data Collection and Processing. pp 114-122. Warrington: SERC Daresbury Laboratory.
Fourm,. R., Shepard, W., Kahn, R., L'Hermite, G. & Li de La Sierra, I. (1995) J. Synchr. Rad. 2, 36-48.
Gilmore, C.J. (1984) J. Appl. Cryst. 17, 42-46.
Grübel, G. (1994) ESRF Beamline Handbook, 55-59.
Harris, G.W. (1995) Acta Cryst. D51, 695-702.
Harrison, S.C. (1969) J. Mol. Biol. 42, 457-483.
Hendrickson, W.A. (1985) Trans. Am. Crystallogr. Assoc. 21, 11-21.
Hütsch, M. (1993) PhD Thesis, University of Hamburg, Germany.
Ibel, K. & Stuhrmann, H.B. (1975) J. Mol. Biol. 93, 255-265.
Jack, A., Harrison, S.C. & Crowther, R.A. (1975) J. Mol. Biol. 97, 163-172.
Kahn, R., Fourme, R., Bosshard, R. & Saintagne, V. (1986) Nucl. Instrum. Methods A246. 596.
Karle, J. (1980) Int. J. Quantum Chem: Quantum Biol. Symp. 7, 357-367
Kortright, J.B. (1986) in "Center for X-ray Optics: X-ray Data Booklet" pp. 2-19 - 2-20, ed.D. Vaughan.
Kühnholz, O. (1991) J. Appl. Cryst. 24, 811-814.
Li de la Sierra. I., Prangé, T., Fourme, R., Padron, G., Fuentes, P., Musacchio A. and Madrazo. J. (1994) J. Mol. Biol. 235, 1154- 1155.
Li de la Sierra, I., Pernot, L., Prangé, T., Saludjian, P., Schiltz, M., Fourme, R. and Padron, G. (1997) J. Mol. Biol., in press.
Lunin, V.Y., Lunina, N.L., Petrova, T.E., Vernoslova, E.A., Urzhumtsev, A.G. & Podjarny, A.D. ( 1995) Acta Cryst. D51, 896-903.
Moras, D., Lorber, B., Romby, P., Ebel, J.-P., Giégé, R., Lewit-Bentley, A. & Roth, M. (1983) J. Biol. Struct. Dynam. 1, 209-223.
Munk, B. (1988) PhD Thesis, University of Hamburg, Germany.
Podjarny, A., Bhat, T.N. & Zwick, M. (1987) Annul Rev. Biophys. Biophys. Chem. 16, 351-374, and references cited therein.
Rey, F., Jenkins, J., Janin, J., Lasters, I., Alard, P., Matthyssens, G. & Wodak, S. (1988) PROTEINS: Structure, Function and Genetics 4, 165-172
Roth, M., Lewit-Bentley, A. & Bentley, G.A. (1984) J. Appl. Cryst. 17, 77-84.
Roth, M. (1991) Crystallographic Computing 5: From Chemistry to Biology, pp 229-248. Oxford University Press. Eds. D. Moras, A.D. Podjarny & J.C. Thierry.
Stuhrmann, H.B. & Kirste, R. (1965) Z. für. Physik. Chem. 46, 247-250.
Stuhrmann, H.B. (1980) J. Appl. Cryst. A36, 996-1001.
Subbiah, S. (1991) Science 252, 128-133.
Suhbbiah, S. (1993) Acta Cryst. D49, 108-119
Urzhumtsev, A.G., Vernoslova, E.A & Podjarny, A.D. (1996) Acta Cryst. D52 1092-1097.
Williams, C.E., May, R.P. & Guinier, A. (1994) in "Materials Science and Technology: A Comprehensive Treatment - Characterisation of Materials Part II" Volume Ed. E. Lifshin, Eds. R.W. Cahn & E.J. Kramer, Vol. 2B, 611-656.