Maximum-Entropy Methods and the Bayesian Programme
G. Bricogne
MRC Laboratory of Molecular Biology, Hills Road, Cambridge CB2 2QH England and LURE, Bâtiment 209D, F-91405 Orsay Cedex, France gb10@mrc-lmb.cam.ac.ukhttp://Lagrange.mrc-lmb.cam.ac.uk
0. Introduction.
The success of direct methods programs at providing a quasi-automatic solution to the phase problem for small molecules has over the years dimmed the perception of the basic inference processes involved in such crystal structure determinations. Greater awareness of this sequence of inference steps has persisted in the macromolecular field, where the dialogue between numerical computation and human decision is still part of the daily experience of most crystallographers. The final step of turning the determination of macromolecular crystal structures itself into a purely computational and automatic process is therefore likely to involve – and even require – that a common basis be found for all phase determination methods used in these two fields. The purpose of this article is to present an overview of one such unifying scheme, the Bayesian programme formulated some years ago by the writer [1,2,3], partial implementations of which have given several encouraging results [4-14] along the way to a full implementation. Special attention is paid here to those areas where this viewpoint is having a practical impact on real applications in macromolecular crystallography. Its application to ab initio phasing at typical macromolecular resolutions will require in addition the incorporation of stereochemical information into structure factor statistics [15].
1. Motivation.
Bayesian concepts and methods are ideally suited to the "management" of crystal structure determination from diffraction data [16]. Indeed, the latter is fundamentally a sequence of steps aimed at gradually reducing the ambiguity created by the loss of phase information. Each step involves the formulation of a range of hypotheses designed to "fill in" the missing information in one way or another, followed by the testing of these hypotheses against the available diffraction data and also against prior chemical knowledge – either as such or after it has been converted into statistical correlations between structure factors.
The work published in [1] was a first step towards this goal, within the restricted context of direct phase determination. Its purpose was to urge a return to the fundamental problem of calculating joint probability distributions of structure factors and to find methods better suited to the macromolecular field which would increase the accuracy and the sensitivity of probabilistic phase indications. Shortcomings of conventional direct methods were identified and shown to be related to the use of uniform distributions of random atomic positions, and of the associated Edgeworth series as an approximation to the joint distribution of structure factors. They were overcome by using instead maximum-entropy distributions [17] for the unknown positions of random atoms, and the associated saddlepoint approximation to the joint probability distribution of structure factors.
The scope of the Bayesian analysis was then enlarged to include other crystallographic methods, particularly those used in the macromolecular field (isomorphous substitution, anomalous scattering, molecular replacement, non-crystallographic symmetry averaging and solvent flattening) whose conventional formulations all involve some form of statistical treatment when they have to deal with the representation of incomplete knowledge – e.g. non-isomorphism in heavy-atom derivatives, or missing atoms in a partial model. These statistical treatments are as a rule rather simplistic in comparison with those on which direct methods are based, and yet it is through the resulting "phase probability densities" that these methods pool their abilities to determine phases. It is therefore no exaggeration to say that macromolecular phasing techniques have so far communicated with each other through their weakest component. These shortcomings were addressed by extending the initial framework into a "multichannel formalism" [2] which made possible the effective construction of a wide range of flexible statistical models involving mixtures of randomly positioned scatterers distributed with varying degrees of non-uniformity. Such models were precisely the hitherto missing devices for optimally describing any kind of phase uncertainty, and it was proposed that the corresponding likelihood functions should be used as a universal tool for consulting data in all conventional macromolecular phasing and refinement methods.
Finally, numerous other classes of situations occur in macromolecular crystallography where the existence of ambiguities is inadequately handled, either by taking centroids of multimodal distributions (as in the Blow & Crick [18] treatment of strongly bimodal phase indications), or by trying to apply iterative map improvement techniques from a single choice of starting point – irrespective of how uncertain that starting point may be (as in solvent flattening from a single choice of molecular boundaries) – creating the risk of potentially disastrous biases. Here again the Bayesian view point leads to a much needed general mechanism for dealing appropriately with ambiguities, which was missing in conventional methods.
2. The Bayesian viewpoint.
The key concept in the Bayesian view of crystal structure determination is the notion of missing information, or ambiguity, in the current situation. Typical instances of missing information encountered in macromolecular crystallography include uncertain molecular boundaries, inconclusive rotation or translation searches, strong bimodality in SIR phase probability indications, and of course the lack of any phase indications whatsoever for some reflexions.
The techniques of Bayesian inference can then be brought into action whenever an item of missing phase information can be shown to influence the expected joint probability distribution of structure factor amplitudes. In every such case, some of the missing information can be retrieved from structure factor amplitudes by the following procedure:
(1) generating an ensemble of hypotheses ![]() forming a "representative sample"
forming a "representative sample" ![]() of all possibilities left open by the current state of ambiguity (to avoid any bias), and assigning to each of them a prior probability
of all possibilities left open by the current state of ambiguity (to avoid any bias), and assigning to each of them a prior probability ![]() from knowledge available outside the diffraction measurements;
from knowledge available outside the diffraction measurements;
(2) constructing the jpd ![]() of the observable structure factors F conditional on each hypothesis
of the observable structure factors F conditional on each hypothesis ![]() ; then integrating the phases out to get the marginal cpd of amplitudes
; then integrating the phases out to get the marginal cpd of amplitudes ![]()
(3) forming the likelihood of each ![]() from the observed data as
from the observed data as
![]() and using Bayes's theorem to obtain the posterior probability of each hypothesis as
and using Bayes's theorem to obtain the posterior probability of each hypothesis as
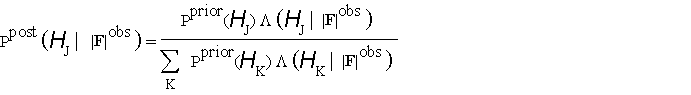 (2.1).
(2.1).
The basic computational mechanism in Bayesian crystal structure determination is therefore :
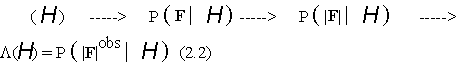
i.e. the conversion of a hypothesis
H into a likelihood function (via a jpd of structure factors) for testing that hypothesis against the available structure factor amplitude data. It is this mechanism which was analysed in [2] for a class of hypotheses wide enough to accommodate all conventional phasing and refinement techniques.3. Basic computational processes.
The techniques involved in implementing the scheme just described fall naturally into three main categories. The first is concerned with the design of efficient sampling strategies for the generation of diverse hypotheses and with the book-keeping of that diversity (§3.1). The second deals with the analytical and numerical aspects of deriving joint probability distributions of structure factors (§3.2), then conditional distribution of amplitudes and likelihood functions (§3.3); these methods are mathematically intensive and can only be outlined here. The third and last category addresses the problem of assessing how much of the initially missing phase information can actually be retrieved from the statistical scores obtained after evaluating all hypotheses in the sample (§3.4).
3.1. Generation Of Diversity: factor permutation.
The generation of a "representative sample" of hypotheses to specify some of the currently missing information may involve a variety of "factors" for which multiple choices remain possible: the unrestricted assignment of trial phase values to totally unphased structure factor amplitudes, or the trial selection of modes if bimodal SIR phase indications pre-exist; choices of plausible molecular boundaries, or of possible redefinitions of an existing boundary; trial placements (i.e. orientations and positions) of plausible molecular substructures; trial definitions of non-crystallographic symmetry elements and/or of the geometric transformations relating multiple crystal forms; and so on. All these factors have in common an ability to influence the expected distribution of the structure factor amplitudes attached to the crystal structure(s) under investigation, and thus a testability against observations of these amplitudes.
Because of the very large number of unknown factors, any scheme for factor permutation has to be hierarchical, which leads unavoidably to a sequential strategy similar to the exploration of moves in a computer chess-playing program. This can be represented by means of a phasing tree, each level of which corresponds to a ply in a chess game. Each node of the tree is a "factor hypothesis", and the early ruling out of some of these hypotheses is reflected by the pruning of the tree.
Call basis set at level
l, denoted Hl , the set of unique reflexions h to which trial phases will be assigned. The hierarchical structure of the search implies that these are nested, i.e. that the basis set grows by concatenating successive increments of new reflexions: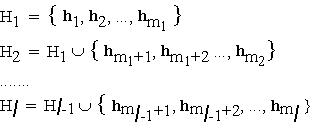
The simplest way to generate phase hypotheses for the reflexions contained in an increment of the basis set is to consider a regular grid of points around each phase circle, e.g. at 45+90k degrees (k=0,...,3) giving a "quadrant permutation" long used in MULTAN [19], or a more general grid. Much more powerful methods exist, based on error-correcting codes, for sampling several phases simultaneously. They are described elsewhere in [3,20].
For mixed factors involving both phases and other factor types, well-known techniques for designing optimal sampling schemes can be used [21,22]. Incomplete factorial designs, first introduced into crystallography by Carter & Carter [23] for the design of crystallisation experiments, were used successfully as general permutation designs for mixed factors involving phases and binary choices of molecular envelope attributes [12,13,14].
3.2. Expression of a phase hypothesis.
This is the mechanism through which the viability of a phase hypothesis, i.e. its structural realisability, is measured. According to the general scheme of §2 two main quantities need to be evaluated, or at least approximated:
(a) the probability that given phased structure factor values ![]() attached to a basis set
attached to a basis set ![]() belong to a chemically valid structure; this function of the F's is called their joint probability distribution and is denoted
belong to a chemically valid structure; this function of the F's is called their joint probability distribution and is denoted ![]() or
or ![]() for short;
for short;
(b) the probability distribution of other structure factor values for a set ![]() of non-basis reflexions over all possible valid structures compatible with the given phased structure factor values in the basis set H ; this is called the conditional probability distribution (cpd) of
of non-basis reflexions over all possible valid structures compatible with the given phased structure factor values in the basis set H ; this is called the conditional probability distribution (cpd) of ![]() given
given ![]() , denoted
, denoted ![]() or
or ![]() for short,. All these probabilities can be calculated, to a consistent degree of accuracy in the whole range of applications, by the maximum-entropy method. The main features of this calculation, described elsewhere [1,2,24] in more detail, may be summarised as follows.
for short,. All these probabilities can be calculated, to a consistent degree of accuracy in the whole range of applications, by the maximum-entropy method. The main features of this calculation, described elsewhere [1,2,24] in more detail, may be summarised as follows.
Let n be a node of the phasing tree at level l , where the basis set Hl has m reflexions, and let ![]() be the vector of phased structure factor values describing the hypothesis attached to this node.
be the vector of phased structure factor values describing the hypothesis attached to this node. ![]() has n real components, where n is the number of centric plus twice the number of acentric reflexions in Hl.
has n real components, where n is the number of centric plus twice the number of acentric reflexions in Hl.
If N identical atoms are thrown at random, independently, with probability density m(x) in the asymmetric unit D, then the saddlepoint approximation [1,2,24] to the prior probability of F(n) is given by:
 where
where ![]() denotes the relative entropy functional
denotes the relative entropy functional
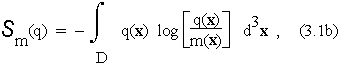
while ![]() denotes the unique distribution compatible with the data in
denotes the unique distribution compatible with the data in ![]() which maximises this relative entropy. Matrix
which maximises this relative entropy. Matrix ![]() is the covariance matrix between the trigonometric structure factor contributions to the components of
is the covariance matrix between the trigonometric structure factor contributions to the components of ![]() when the random atoms are distributed with density
when the random atoms are distributed with density ![]() ; it is calculated by structure factor algebra [1,2,24].
; it is calculated by structure factor algebra [1,2,24].
3.3. Assay of phase hypotheses.
To measure the "strength of binding to the data" of a phase hypothesis, we will try and assess to what extent that hypothesis is able to guess some characteristics of the distribution of data it has not yet seen – an idea which bears some similarity to that of cross-validation.
For this purpose we rely on maximum-entropy extrapolation as a prediction mechanism for structure factors: besides reproducing the amplitudes and phases ![]() attached to node n of the phasing tree for reflexions in the basis set Hl , the maximum-entropy distribution
attached to node n of the phasing tree for reflexions in the basis set Hl , the maximum-entropy distribution ![]() also gives rise to Fourier coefficients
also gives rise to Fourier coefficients ![]() with non-negligible amplitude for many non-basis reflexions, i.e. for k in the complement Kl of Hl . This phenomenon is known as maximum-entropy extrapolation. It is the Bayesian equivalent of the tangent formula of conventional direct methods (see §3.4 of [1] for a more detailed discussion). Intuitively, the maximum-entropy extrapolate
with non-negligible amplitude for many non-basis reflexions, i.e. for k in the complement Kl of Hl . This phenomenon is known as maximum-entropy extrapolation. It is the Bayesian equivalent of the tangent formula of conventional direct methods (see §3.4 of [1] for a more detailed discussion). Intuitively, the maximum-entropy extrapolate ![]() is that value of
is that value of ![]() which can be fitted "for free" once the basis-set constraints
which can be fitted "for free" once the basis-set constraints ![]() have been fitted.
have been fitted.
The conditional distribution ![]() is affected by this extrapolation, since we may write:
is affected by this extrapolation, since we may write:
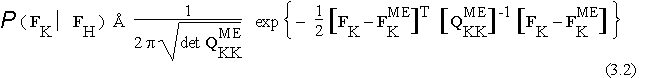
and so will the (marginal) conditional distribution of amplitudes ![]() obtained by integrating over the phases present in FK . We will therefore obtain a prediction of the distribution of
obtained by integrating over the phases present in FK . We will therefore obtain a prediction of the distribution of ![]() which depends on the phases attached to node n . The likelihood
which depends on the phases attached to node n . The likelihood ![]() of the trial phases
of the trial phases ![]() in
in ![]() is the conditional probability of the observed values for the amplitudes
is the conditional probability of the observed values for the amplitudes ![]() :
:
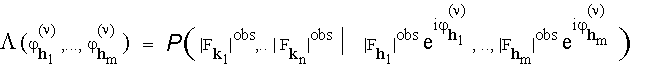
It is often convenient to consider the log-likelihood ![]() . Let (
. Let (![]()
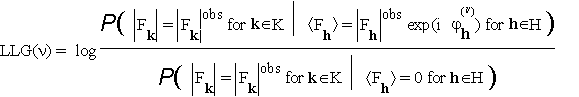 (3.4).
(3.4).
This quantity will be largest when the phase assumptions attached to node ![]() lead one to expect deviations from Wilson statistics for the unphased amplitudes
lead one to expect deviations from Wilson statistics for the unphased amplitudes ![]() , k
, k![]() K, that most closely match those present in the distribution of the actual measurements
K, that most closely match those present in the distribution of the actual measurements ![]() . It is therefore a quantitative measure of the degree of corroboration by the unphased data of the phase assumptions attached to
. It is therefore a quantitative measure of the degree of corroboration by the unphased data of the phase assumptions attached to ![]() .
.
The most fundamental likelihood function is that of Rice[25], derived as the marginal distribution for the amplitude R of an offset 2D Gaussian:
![]() (3.5a)
(3.5a)
in which r is the offset length and ![]() the variance parameter. Typically r is the modulus of an Fcalc, R is the observed modulus
the variance parameter. Typically r is the modulus of an Fcalc, R is the observed modulus ![]() and the variance parameter
and the variance parameter ![]() is derived e.g. by Wilson statistics to represent the statistical dispersion of a contribution from random atoms.
is derived e.g. by Wilson statistics to represent the statistical dispersion of a contribution from random atoms.
It is straightforward, starting from a 1D rather than 2D Gaussian, to obtain the centric equivalent of the Rice distribution as:
![]() (3.5b).
(3.5b).
The derivation of both likelihood functions assumes that all phases or signs over which integration is carried out have equal probability. In the acentric case it is further assumed that the original 2D Gaussian is isotropic. The first assumption is invalid when prior phase information is available from MIR or MAD, while the second is violated when the distribution of random atoms is strongly non-uniform or obeys non-crystallographic symmetries. Fortunately both of these generalisations can be carried out simultaneously by defining the elliptic Rice distribution (although Rice never considered it) required for a "phased LLG", which was derived in [26].
As a decision criterion likelihood enjoys certain optimality properties both in conventional statistics (Neyman-Pearson theorem) and of course in Bayesian statistics through its role in Bayes's theorem (2.1). In both settings, likelihood evaluation or optimisation is the common mechanism through which the Bayesian structure determination process interrogates the observed data for the purpose of testing or refining hypotheses. The examples given in sections 4 to 7 provide an ample illustration of the confluence of a wide range of hitherto distinct detection and refinement operations into a single calculation, namely computing value and derivatives for a suitable likelihood function.
Whenever likelihood does not totally dominate prior knowledge, the full force of Bayes's theorem should be invoked. Using Bayes's theorem in the form (2.1) together with expression (3.1a,b) for the prior probability of a set of trial structure factor values, the a-posteriori probability Ppost(![]() ) of the phase hypothesis attached to node
) of the phase hypothesis attached to node ![]() of the phasing tree may be evaluated by first computing the Bayesian score:
of the phasing tree may be evaluated by first computing the Bayesian score:
![]() (3.6)
(3.6)
(where N is the number of independent atoms in the asymmetric unit) and exponentiating it, then normalising this collection of numbers over a suitable collection of nodes ![]() .
.
3.4. Analysis of phase hypotheses.
In the course of the tree-directed search described above, a subpopulation of nodes with high scores will progressively be selected. Rather than consider this list of instances as the end product of the phasing process, we want to do some data reduction and relate the property of achieving these high scores to the property of having the right values for some critical phases or combinations of phases (or other factors).
Since the initial sampling plan according to which the nodes were generated (§3.1) will normally be based on some efficient sampling design, this is a typical setting in which to call upon standard techniques for analysing the results of "designed experiments" [27-30].
Phases, however, are special factors because of their periodic character. As a result any phase-dependent score function, for instance the Bayesian score B = B (![]() ), is a periodic function, with an m-dimensional period lattice generated by translations of 2
), is a periodic function, with an m-dimensional period lattice generated by translations of 2
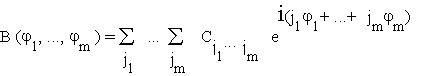 .
.
We are looking for those C![]() which are significantly higher in the successful nodes that in the general population, i.e. higher than one would expect in the absence of any trends. The issues of optimal sampling and of statistical analysis of node scores therefore belong to the realm of multidimensional Fourier analysis [3,20] . For factors other than phases, which may not be periodic, conventional techniques such as multiple linear regression [27] may be used.
which are significantly higher in the successful nodes that in the general population, i.e. higher than one would expect in the absence of any trends. The issues of optimal sampling and of statistical analysis of node scores therefore belong to the realm of multidimensional Fourier analysis [3,20] . For factors other than phases, which may not be periodic, conventional techniques such as multiple linear regression [27] may be used.
Standard methods are available for assessing the level of significance of the results of a multiple regression analysis of experimental scores [27]. The simplest of them, namely the application of Student’s t-test to the determination of a single sign, was described in §2.2.4 of [3] and was used [31] in the solution of a powder structure.
4. Overview of selected applications.
In the Bayesian picture, the privileged role played by the Rice likelihood functions in consulting the experimental observations leads naturally to delineating three main "regimes" in the process of structure determination, corresponding to three distinct approximation regimes for these functions: a "Patterson correlation regime", a "transition regime" and a "Fourier correlation regime". A number of examples pertaining to each regime will be worked out in some detail in the forthcoming sections. It will be a recurring observation that the Bayesian analysis turns out to yield improvements of these methods which had not (or had only just) arisen within their own theoretical framework, at the same time as providing an automatic unification of these improvements within a common computational mechanism.
4.0. Approximation regimes for the Rice log-likelihood functions.
We will begin with a few basic results on Wilson statistics and associated notation. For h acentric, F(h) is distributed as a 2D Gaussian centred at (0,0) with variance
![]() (4.0a)
(4.0a)
along each component, while for h centric it is distributed as a 1D Gaussian centred at 0 with variance
![]() (4.0b)
(4.0b)
where
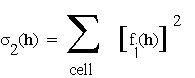 . (4.0c)
. (4.0c)
It will also be recalled, for later use, that standard normalised structure factor amplitudes are defined by
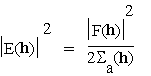 for h acentric (4.0d)
for h acentric (4.0d)
and 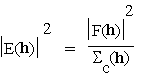 for h centric (4.0e)
for h centric (4.0e)
so that ![]() for all h (4.0f).
for all h (4.0f).
Define the acentric and centric Rice log-likelihood functions La and Lc as the logarithms of (3.5a) and (3.5b) using variances Sa and Sc respectively, and introduce the following shorthand for the Sim figures of merit:
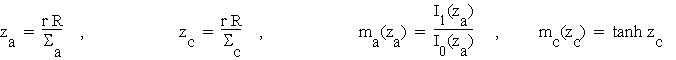 (4.1)
(4.1)
where I0 and I1 are the modified Bessel functions of order 0 and 1 respectively. It is then straightforward to obtain the following partial derivatives:
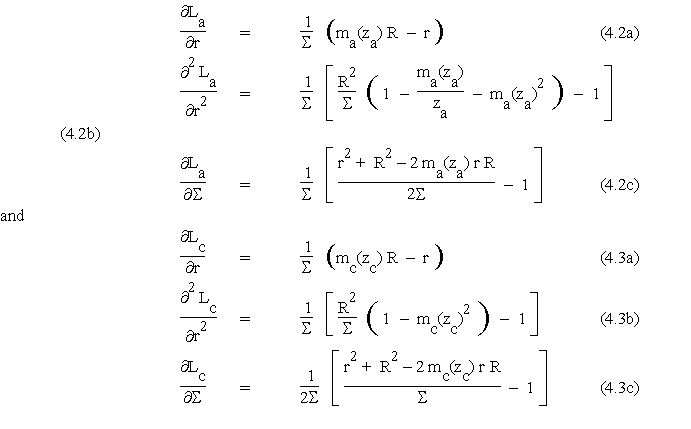 which will be used to build Taylor expansions of La and Lc for small variations of r and S .
which will be used to build Taylor expansions of La and Lc for small variations of r and S .
When the quantity z in (4.1) becomes large, the asymptotic formulae
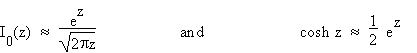 (4.4a,b)
(4.4a,b)
give rise to the following approximations:
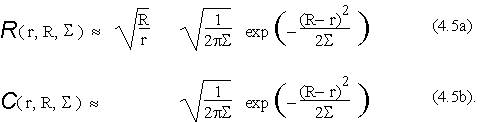
4.1. The Patterson correlation regime.
In detection problems the standard situation is that a null hypothesis with vanishing offsets r and some initial variances ![]() is to be compared to alternative hypotheses with non-zero offsets dr and lower variances
is to be compared to alternative hypotheses with non-zero offsets dr and lower variances ![]() , where
, where ![]() is related
is related ![]() through Wilson statistics.
through Wilson statistics.
For r = 0 we have ma(za) = mc(zc) = 0 so that the first-order derivatives (4.2a) and (4.3a) vanish. Using the limiting value 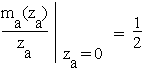 and the basic relations (4.0a-f) we get the local Taylor expansions :
and the basic relations (4.0a-f) we get the local Taylor expansions :
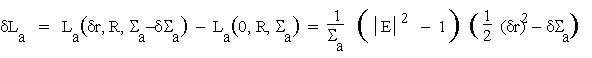 (4.6a)
(4.6a)
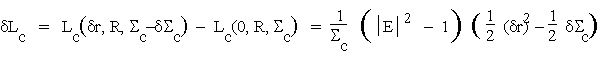 (4.6b)
(4.6b)
When summed over all reflexions these quantities will be shown to give rise to correlation functions between the origin-removed ![]() -based Patterson function for the observed data and the origin-removed Patterson for the partial structure whose presence is to be detected.
-based Patterson function for the observed data and the origin-removed Patterson for the partial structure whose presence is to be detected.
Once a partial structure has been detected, subsequent searches for more partial structures can be carried out with the benefit of the phase information generated by the first: one has then entered the "transition regime" (§4.2 below).
4.2. The transition regime.
The transition regime applies when the offsets r are no longer zero but the quantities z in (4.1) are small (a few units at most). The first-order derivatives (4.2a) and (4.3a) no longer vanish and hence create in the Taylor expansion of La or Lc an incipient Fourier-like sensitivity to the existing phase information. However the second-order terms remain substantial and continue contributing Patterson-like features. The simple proportionality relations between 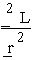 and
and ![]() for r=0 which give rise to (4.6a,b) cease to hold: the
for r=0 which give rise to (4.6a,b) cease to hold: the ![]() associated to these two types of partial derivatives are renormalised differently under the available phase information.
associated to these two types of partial derivatives are renormalised differently under the available phase information.
This is the "middle game" situation, where phase information is beginning to emerge but remains highly ambiguous. A large fraction of the observed intensities has to be accounted for through variance rather than expectation, reflecting the large amount of missing phase information. A useful measure of missing phase information at a reflexion h is the renormalised ![]() associated to
associated to ![]() , which may be written
, which may be written
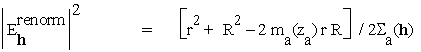 for h acentric (4.7a)
for h acentric (4.7a)
= ![]() for h centric (4.7b).
for h centric (4.7b).
This quantity was considered by Nixon & North [32]. Reflexions h for which it is the largest are those where phase information is "most sorely missing", and hence are the best candidates for phase permutation (§3.1), or for the permutation of any other factor capable of leading to a lower renormalised ![]() . This selection procedure was used successfully in the statistical phasing of Tryptophanyl-tRNA synthetase [12,13,14].
. This selection procedure was used successfully in the statistical phasing of Tryptophanyl-tRNA synthetase [12,13,14].
4.3. The Fourier correlation regime.
This regime is reached when most of the quantities z in (4.1) have appreciable values (several units). According to (4.5a,b) the likelihood function becomes approximately that of a least-squares residual on amplitudes, but with the important feature that the variances may still come mostly from the structure factor statistics rather than from the observational errors.
Current structure refinement protocols (PROLSQ [33], TNT [34] and XPLOR [35]) are still based on a least-squares residual, hence ignore this extra source of variance. As a result, they suffer from well-known problems of model bias. Maximum-likelihood structure refinement is proposed in §7.2 as a superior alternative to least-squares refinement, and this claim is supported by encouraging test results.
4.4. Introduction to the examples.
The selection of illustrations given in the forthcoming sections comprises a varied range of applications taken from both macromolecular and direct methods techniques. In the latter, the Bayesian viewpoint brings simplicity and clarity to what has traditionally been a thicket of formulae, and enlarges both their scope and their effectiveness. In the former, it also provides a thread of continuity between hitherto distinct techniques and reveals numerous possibilities of substantial improvements. Most importantly, this survey demonstrates the extent to which all existing techniques are subsumed within a unique computational protocol which needs no longer be aware of the multitude of specialisations through which it has so far been approximated.
5. Application to detection problems.
5.1. Heavy-atom detection in a structure.
In the standard use of the method for small molecules the "heavy atom" is first detected by examination of the Patterson, taking advantage of the fact that such an atom is localised and poses no rotation problem. One then switches over to Sim's formula [36] which can be expected to hold rather accurately since the 'light' atoms making up the rest of the structure are distributed uniformly enough for Wilson's statistics to be obeyed (small-molecule crystals are usually close-packed – there is no solvent – and the exclusion of light atoms by the heavy atom can be neglected in most cases).
The statistical treatment of the same problem shows readily that the optimal detection function under the "Patterson regime" approximation [26,37] has the form of a Patterson correlation (PC) function [38] between the origin-removed ![]() –based Patterson for the whole structure and the heavy-atom origin-removed Patterson.
–based Patterson for the whole structure and the heavy-atom origin-removed Patterson.
This approximation is valid only if the light atoms are distributed so as to give rise to Wilson statistics; it may be known that this is not the case, e.g. in crystals of zeolites or other small structures containing cavities, and of course in macromolecular crystals. A full LLG evaluation has no difficulty in remaining an optimal detection criterion in this case, while the PC coefficient will fail to do so. Furthermore the statistical variances used can be incremented so as to reflect measurement errors, while there is no natural way of doing so when calculating the PC coefficient.
5.2. Heavy-atom detection in the MIR and MAD methods.
The statistical theory of heavy-atom parameter refinement, and the SHARP program implementing it, are dealt with in [39]. However the problem of statistical heavy-atom detection is best illustrated by reference to analytical formulae for SIR likelihood functions derived in [40].
Statistical detection begins with a maximum-likelihood estimation of scale factors and of non-isomorphism ![]() under the null hypothesis that all discrepancies between
under the null hypothesis that all discrepancies between ![]() caused by non-isomorphism. The alternative hypotheses assume instead the presence of a "nascent" heavy atom at xH whose occupancy increases from 0 while
caused by non-isomorphism. The alternative hypotheses assume instead the presence of a "nascent" heavy atom at xH whose occupancy increases from 0 while ![]() decreases by the corresponding amount as in §5.1.2. Approximating LLG(xH) by a second order Taylor expansion [26] yields a detection criterion of the form of a Patterson correlation function involving quantities which can be written as
decreases by the corresponding amount as in §5.1.2. Approximating LLG(xH) by a second order Taylor expansion [26] yields a detection criterion of the form of a Patterson correlation function involving quantities which can be written as ![]() and may be recognised as the normalised (sharpened) and origin-removed versions of coefficients advocated by Kalyanaraman & Srinivasan [41] as being the best ones from which to compute a difference-Patterson function for the determination of heavy-atom positions using isomorphous data. Preliminary tests in SHARP have shown that this criterion is indeed very promising for weak isomorphous signals, and a generalisation of the Rice distibution is being developed for detecting anomalous scatterers from MAD data sets.
and may be recognised as the normalised (sharpened) and origin-removed versions of coefficients advocated by Kalyanaraman & Srinivasan [41] as being the best ones from which to compute a difference-Patterson function for the determination of heavy-atom positions using isomorphous data. Preliminary tests in SHARP have shown that this criterion is indeed very promising for weak isomorphous signals, and a generalisation of the Rice distibution is being developed for detecting anomalous scatterers from MAD data sets.
5.3. Fragment detection in the molecular replacement method.
Instead of a heavy atom as in §5.1 we now have a known fragment described in a reference position and orientation by a density ![]() with transform FM . If
with transform FM . If ![]() is rotated by R and translated by t to give the copy of the fragment lying in the chosen asymmetric unit, then statistical detection and placement of the fragment will proceed by calculating the log-likelihood gain
is rotated by R and translated by t to give the copy of the fragment lying in the chosen asymmetric unit, then statistical detection and placement of the fragment will proceed by calculating the log-likelihood gain
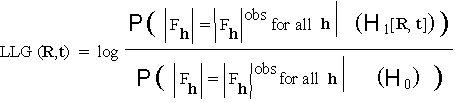 (5.1)
(5.1)
where (![]() ) denotes the null hypothesis that all atoms (including those of the fragment) are uniformly distributed in the asymmetric unit while (
) denotes the null hypothesis that all atoms (including those of the fragment) are uniformly distributed in the asymmetric unit while (![]() ) denotes the alternative hypothesis that a subset of atoms is assembled into the known fragment and placed in the asymmetric unit with orientation R at position t , and the rest are distributed at random.
) denotes the alternative hypothesis that a subset of atoms is assembled into the known fragment and placed in the asymmetric unit with orientation R at position t , and the rest are distributed at random.
The methods used in §5.1 then carry over to the present situation and yield [26,42] a detection criterion consisting of two terms. The first term depends only on the rotational placement R and is a PC-based rotation function in which a sum of point-group symmetry-related copies of the origin-removed self-Patterson of the rotated fragment is being correlated with the origin-removed sharpened self-Patterson of the whole structure. The second term, considered for a fixed value of the rotational component R of the placement, gives rise to a PC-based translation function, expressed as a Fourier series. The fact that the log-likelihood gain (which is an optimal criterion by the Neyman-Pearson theorem is based on E's provides a final explanation to the long-standing observations by Ian Tickle [42] that E-based (sharpened) translation functions always give better results than F-based (unsharpened) ones.
It is therefore clear in this case too that even the most approximate implementation of the statistical detection approach yields better criteria than the most sophisticated ones available so far, and suggests non-trivial improvements of the existing methodology which had not yet arisen within this methodology itself.
5.4. Detection of non-uniformity from variance modulation.
In small-molecule direct methods the phase determination process is often primed by means of so-called S1 relations, which give immediate estimates of certain phases belonging to a subclass of reflexions (see e.g. [43]). Giacovazzo [44,45] and Pavel ík [46,47] have found a connection between these relations and Harker sections in Patterson functions.
It was shown in [26] that ![]() relations are related to the sensitivity of the Rice log-likelihood to its variance parameter and to the modulation of the latter by the non-uniformity of atomic distributions. Thus
relations are related to the sensitivity of the Rice log-likelihood to its variance parameter and to the modulation of the latter by the non-uniformity of atomic distributions. Thus ![]() relations are a purely variance-based method for detecting non-uniform distribution of all atoms, by the same statistical technique (likelihood-based hypothesis testing) which was used earlier for detecting the non-uniform distribution of one heavy atom in a background of light-atoms.
relations are a purely variance-based method for detecting non-uniform distribution of all atoms, by the same statistical technique (likelihood-based hypothesis testing) which was used earlier for detecting the non-uniform distribution of one heavy atom in a background of light-atoms.
6. Application to completion problems.
6.1. Detection of further heavy atoms or fragments.
All detection problems in sections 5.1 to 5.3 were examined under the assumption that no phase information existed (there is no basis set), so that the ordinary Rice likelihood functions (3.18a,b) apply. If we now assume that some external phase information has become available, the elliptic Rice likelihood function [26] should be used instead. If this phase information is strong and unimodal, then the LLG is essentially an electron density correlation function (a "phased rotation/translation function") as discussed in §4.3. If not, the LLG will possess features intermediate between those of a Patterson correlation coefficient and of a density correlation coefficient, giving rise to what might be called partially phased rotation and translation functions.
The log-likelihood gradient maps used in SHARP (§7.1 and [39]) are based on this idea. They have proved highly successful in revealing fine details of heavy-atom substitution such as minor sites, anisotropy of thermal disorder, and split sites caused by multiple conformations.
6.3. Maximum-entropy solvent flattening.
When a great deal of phase information is specified in H and the prior prejudice m(x) is non-uniform, the expressions above are of little help but numerical computation can proceed unimpeded. The first successful use of this phase extension procedure was reported in [11]. It showed on the cytidine deaminase structure that maximum-entropy solvent flattening (MESF) using the solvent mask as a non-uniform prior prejudice provided a better method of phase extension than did ordinary solvent flattening [48]. An interesting aspect of this work lies in the protocol used to build the maximum-entropy distribution when the basis-set phase information consists of MIR phases and is therefore tainted with noise. The preferred way of fitting such constraints would be to aim for the maximum Bayesian score (3.6), but this would require knowing the value N of the number of atoms in the structure, a quantity which is difficult to define at non-atomic resolution [1]. Instead, the fitting the noisy constraints was allowed to proceed only as long as the LLG outside the basis set kept increasing, and was halted when the LLG reached a maximum. In this way, only that part of the noisy constraints was fitted which contains the ‘signal’, i.e. which improves the predictive power of the statistical model. This is the familiar idea of cross-validation, and in fact this procedure carries out something akin to an estimation of the "effective N" by cross-validation, as well as cross-validated density modification by exponential modelling.
6.4. Hypothesis permutation in the 'middle game' of structure determination.
The crystal structure determination of the tetragonal form of Bacillus Stearothermophilus tryptophanyl-tRNA synthetase (TrpRS) provided the first application to the determination of an unknown macromolecuar structure of the full Bayesian scheme (§2) for inferring missing phase information.
Unlike the case of cytidine deaminase on which the MESF procedure was first tested (§6.3), the case of TrpRS was marred by serious non-isomorphism in the heavy-atom derivatives, resulting in large starting-phase errors and hence in a poor definition of the molecular envelope. MESF proved unable to produce better maps from such an unfavourable starting point: instead it led to a severe deterioration of the maps, accompanied by a dramatic decrease of the LLG statistic as phases were extended from about 5.0Å resolution to the 2.9Å limit of the diffraction data. It was therefore necessary to somehow improve simultaneously the quality of the starting phases and the correctness of the molecular envelope - a task whose circularity from the conventional standpoint made it at first sight as impossible as lifting oneself by one’s bootstraps.
The deadlock was broken by a straightforward application of the exploratory process described in §2. The "most sorely missing information" was found to be associated with strong reflexions having large renormalised ![]() values (§4.2) which were initially unphased and were inaccessible by maximum-entropy extrapolation from the phased ones. Their phases were permuted, at first on their own, then together with permuted hypotheses concerning possible modifications of the molecular envelope. All permutations were carried out by using incomplete factorial designs [23]. For each such specification of the new phases and of the envelope the MESF process was applied, the maximal value reached by the LLG statistic was noted, and these scores were subsequently analysed by multiple-regression least-squares. Student t-tests were performed to assess significance, and turned out to provide reliable indications for most of the phases of 28 strong reflexions and for the six binary choices of envelope attributes involved in the permutations. The resulting phase improvement made it possible to assign positions, hitherto unobtainable, for nine of the ten selenium atoms in an isomorphous difference Fourier map for SeMet-substituted TrpRS. Further phase permutation continued to produce improved maps from the pooled MIR phase information and played a critical role in solving the structure [12]. This is the first practical demonstration of the effectiveness of the Bayesian approach at a typical macromolecular resolution [14].
values (§4.2) which were initially unphased and were inaccessible by maximum-entropy extrapolation from the phased ones. Their phases were permuted, at first on their own, then together with permuted hypotheses concerning possible modifications of the molecular envelope. All permutations were carried out by using incomplete factorial designs [23]. For each such specification of the new phases and of the envelope the MESF process was applied, the maximal value reached by the LLG statistic was noted, and these scores were subsequently analysed by multiple-regression least-squares. Student t-tests were performed to assess significance, and turned out to provide reliable indications for most of the phases of 28 strong reflexions and for the six binary choices of envelope attributes involved in the permutations. The resulting phase improvement made it possible to assign positions, hitherto unobtainable, for nine of the ten selenium atoms in an isomorphous difference Fourier map for SeMet-substituted TrpRS. Further phase permutation continued to produce improved maps from the pooled MIR phase information and played a critical role in solving the structure [12]. This is the first practical demonstration of the effectiveness of the Bayesian approach at a typical macromolecular resolution [14].
The use of the renormalised ![]() value as a criterion for choosing candidates for phase permutation bears an interesting relationship to the expression giving the mean-square noise level in a centroid map as a function of the distribution of figures of merit [49,50,51]. Indeed
value as a criterion for choosing candidates for phase permutation bears an interesting relationship to the expression giving the mean-square noise level in a centroid map as a function of the distribution of figures of merit [49,50,51]. Indeed  is closely analogous to the quantity
is closely analogous to the quantity ![]() , where mh is the figure of merit [18], which gives the contribution of h to the overall noise in the centroid map. Permuting the phases of reflexions having the largest is therefore the fastest way to "remove heat from the system". Acentric reflexions of this type fall into two distinct categories, according to the character of their phase probability densities: (1) those for which it is flat , in which case phase permutation has to proceed in the same way as in ab initio phasing; (2) those for which it is bimodal, for which it is preferable to use mode permutation which boils down to a simple binary choice. In the latter case simultaneous choices of modes for several reflexions may be sampled efficiently by invoking the combinatorial techniques described in [20] These multiple choices may then be evaluated by means of the elliptic Rice likelihood [26] which measures the extent to which the phases extrapolated from each combination of binary choices of modes in the basis set agree with one of the modes for each second neighbourhood reflexion.
, where mh is the figure of merit [18], which gives the contribution of h to the overall noise in the centroid map. Permuting the phases of reflexions having the largest is therefore the fastest way to "remove heat from the system". Acentric reflexions of this type fall into two distinct categories, according to the character of their phase probability densities: (1) those for which it is flat , in which case phase permutation has to proceed in the same way as in ab initio phasing; (2) those for which it is bimodal, for which it is preferable to use mode permutation which boils down to a simple binary choice. In the latter case simultaneous choices of modes for several reflexions may be sampled efficiently by invoking the combinatorial techniques described in [20] These multiple choices may then be evaluated by means of the elliptic Rice likelihood [26] which measures the extent to which the phases extrapolated from each combination of binary choices of modes in the basis set agree with one of the modes for each second neighbourhood reflexion.
6.6 Outlook.
If the expected gains in sensitivity of detection (§5.3), in efficiency of recycling (§6.2) and in effectiveness of completion (§6.4-5) brought about by the full implementation of the Bayesian approach actually materialise, it would become conceivable to attack the ab initio determination of protein structures by systematically searching for super-secondary fragments of 20 to 30 amino-acids, for which it may be possible to compile a library similar to the library of short fragments used for assisting map interpretation by Jones & Thirup [52]. If this does not work without some startup phase information, then an initial round of phase permutation may afford a means of building up enough phase information ab initio in order to increase the detection sensitivity for such fragments above a critical threshold.
This line of development is connected to the more radical approach presented in [15] where the stereochemical information pertaining to such libraries of fragments is incorporated directly into the structure factor statistics from which joint probabilities are built.
7. Application to refinement problems.
7.1. Maximum-Likelihood heavy-atom refinement (SHARP).
This topic is treated in a separate contribution to this Volume. The main difference between least-squares (LS) and maximum-likelihood (ML) parameter refinement resides in the fact that one integrates the partial derivatives around the native phase circle, just as one integrates the structure factor in the Blow & Crick method [18]. This removes the bias previously introduced by phase "estimates" [53] which were particularly questionable in the SIR method. A more subtle difference is that this integration is carried out also in the radial direction to deal with the measurement errors on native amplitudes, and with the absence of a native measurement in the MAD method. Last but not least, the ML method also allows the refinement of parameters describing the lack of isomorphism and hence influencing the weighting of observations; such a refinement is impossible within the least-squares method where weights are necessarily assumed to be fixed.
The rapidly growing list of SHARP successes and the remarkable sensitivity of its LLG gradient maps demonstrate that the full implementation of the ML method for heavy-atom parameter refinement was well worth the extra effort it required.
7.2. Maximum-Likelihood structure refinement.
The Bayesian viewpoint has long suggested that structure refinement should be carried out by maximising the LLG rather than by minimising the conventional least-squares residual [2,7,37]. Here again, only the ML method can take into account the uncertainty of the phases associated to model incompleteness and imperfection by suitably downweighting the corresponding amplitude constraints. It was predicted [37] that ML refinement would allow the refinement of an incomplete model by using the structure factor statistics of randomly distributed scatterers to represent the effects of the missing atoms, in such a way that the latter would not be wiped out; and that the final LLG gradient map would then provide indications about the location of these missing atoms.
These predictions have now been confirmed by actual tests. Bricogne & Irwin have used BUSTER and TNT on a test data set for crambin [54] suffering from both model imperfection and model incompleteness, and compared the results of LS and ML refinements from these data [55]. In these conditions ML refinement clearly outperformed LS refinement, giving a mean-square distance to the correct positions of 0.176 (ML) instead of 0.415 (LS). Furthermore the final LLG gradient map produced by the ML method showed highly significant, correct connected features for the missing part (40%) of the molecule, while the final LS difference map showed no such features. This enhances the possibilities of "bootstrapping" from an otherwise unpromising molecular replacement starting point to a complete structure. Essentially the same behaviour was observed at 2.0Å resolution, and with experimental rather than calculated data.
Other prototypes for ML structure refinement have been built and tested by Read [56] (using XPLOR and an intensity-based LLG) and by Morshudov [57] (using PROLSQ and the Rice LLG). The BUSTER+TNT prototype has the advantage of being able to use external phase information by means of the elliptic Rice function (see [26]), as well as prior information about non-uniformity in the distribution of the missing atoms in incomplete models. It also allows the ML refinement of an incomplete model to be carried out in conjunction with phase permutation or phase refinement for those strong amplitudes which are most poorly phased by that model, i.e. have the largest renormalised ![]() 's ; or in conjunction with maximum-entropy updating of the distribution of random atoms, initially taken as essentially featureless within the given envelope. Using the method of joint quadratic models of entropy and LLG described in [1] before and after refinement of the incomplete model produced updated ME distributions showing the missing structure in its entirety, demonstrating clearly the advantage of carrying out ML refinement within the integrated statistical framework provided by BUSTER. This ME "after-burner" establishes a seamless continuity between the middle game of structure determination and the end game of structure refinement.
's ; or in conjunction with maximum-entropy updating of the distribution of random atoms, initially taken as essentially featureless within the given envelope. Using the method of joint quadratic models of entropy and LLG described in [1] before and after refinement of the incomplete model produced updated ME distributions showing the missing structure in its entirety, demonstrating clearly the advantage of carrying out ML refinement within the integrated statistical framework provided by BUSTER. This ME "after-burner" establishes a seamless continuity between the middle game of structure determination and the end game of structure refinement.
8. Conclusion.
As shown by the current applications, ranging from ab initio phasing through structure completion to structure refinement, all aspects of the determination of crystal structures are inextricably linked to a common body of statistical methods and concepts. The goal of the "Bayesian programme" is to invite a comprehensive implementation of these methods into an integrated software system for users of crystallographic phasing techniques in the macromolecular field.
An appreciation of the benefits which can be expected to follow from this systematic approach can be obtained by recalling that, a very short while ago, it would have seemed insane to even consider using experimental heavy-atom phases in macromolecular structure refinement: heavy-atom phasing still produced severely biased results, creating numerous carbuncles in electron-density maps; and structure refinement suffered from the biases of the least-squares method which would have further amplified the ugly effects of these phase errors. Today, ML heavy-atom refinement with SHARP delivers safe experimental phase probability densities; and ML structure refinement using for example BUSTER+TNT with the elliptic Rice likelihood function can now safely exploit this phase information to widen the domain of convergence of the refinement.
Finally it should be clear that the statistical analysis at stage 0 of the basic procedure in §2, which allows one to identify the "most sorely missing phase information" for the purpose of seeking to obtain it computationally, could equally well be invoked dynamically for deciding how to obtain it experimentally in the most effective way. Such a procedure of "Phasing on the beamline" would extend the field of use of statistical methods into the realm of experimental strategy.
Acknowledgements.
I thank my colleagues in the BUSTER Development Group (John Irwin, Eric de La Fortelle and Pietro Roversi) for their numerous contributions towards the realisation of the Bayesian programme.
This research was supported in part by a Tage Erlander Guest Professorship from the Swedish Natural Sciences Research Council (NFR) in 1992-93, by an International Research Scholars award from the Howard Hughes Medical Institute since 1993, and by grants from Pfizer Inc. and Glaxo-Wellcome PLC since 1996.
References.
[1] G. Bricogne, Acta Cryst. A40, 410-445 (1984).
[2] G. Bricogne, Acta Cryst. A44, 517-545 (1988).
[3] G. Bricogne, Acta Cryst. D49, 37-60 (1993).
[4] G. Bricogne and C.J. Gilmore, Acta Cryst. A46, 284-297 (1990).
[5] C.J. Gilmore, G. Bricogne, and C. Bannister, Acta Cryst. A46, 297-308 (1990)..
[6] C.J. Gilmore and G. Bricogne, this Volume.
[7] G. Bricogne, Acta Cryst. A47, 803-829 (1991).
[8] C.J. Gilmore, K. Henderson, and G. Bricogne, Acta Cryst. A47, 830-841 (1991).
[9] C.J. Gilmore, A.N. Henderson, and G. Bricogne,. Acta Cryst. A47, 842-846 (1991).
[10] W. Dong, T. Baird, J.R. Fryer, C.J. Gilmore, D.D. MacNicol, G. Bricogne, D.J. Smith, M.A. O'Keefe and S. Hovmöller, Nature 355, 605-609 (1992).
[11] S. Xiang, C.W. Carter Jr., G. Bricogne and C.J. Gilmore (1993). Acta Cryst. D49, 193-212.
[12] S. Doublié, S. Xiang, C.J. Gilmore, G. Bricogne and C.W. Carter Jr. Acta Cryst. A50, 164-182 (1994).
[13] S. Doublié, G. Bricogne, C.J. Gilmore and C.W. Carter Jr. (1995). Structure 3, 17-31.
[14] C.W. Carter Jr. (1995). Structure 3, 147-150.
[15] G. Bricogne, Methods in Enzymology 277, in the press.
[16] S. French, Acta Cryst. A34, 728-738 (1978).
[17] E.T. Jaynes, Papers on Probability, Statistics and Statistical Physics, edited by R.D. Rosenkrantz, D. Reidel Publishing Co, Dordrecht (1983).
[18] D.M. Blow and F.H.C. Crick, Acta Cryst. 12, 794-802 (1959).
[19] G. Germain and M.M. Woolfson, Acta Cryst. B24, 91-96 (1968).
[20] G. Bricogne, Methods in Enzymology, 276, 424-448.
[21] W.G. Cochran and G.M. Cox, Experimental Designs, 2nd edition, John Wiley and Sons, New York (1957).
[22] A.C. Atkinson and A.N. Donev, Optimum Experimental Designs, Clarendon Press, Oxford (1992).
[23] C.W. Carter Jr. and C.W. Carter, J. Biol. Chem. 254, 12219-12223 (1979).
[24] G. Bricogne, In Maximum Entropy in Action, edited by B. Buck and V.A. Macaulay, 187-216, Oxford University Press, Oxford (1991).
[25] S.O. Rice, Bell System Tech. J. 23, 283-332 (parts I and II) ; 24, 46-156 (parts III and IV) (1944, 1945). Reprinted in Selected Papers on Noise and Stochastic Processes, ed. by N. WAX, 133-294, Dover Publ., New York (1954).
[26] G. Bricogne, Methods in Enzymology, 276, 361-423.
[27] R.H. Myers, Classical and Modern Regression with Applications, 2nd edition. PWS-KENT, Boston (1986).
[28] R. Mead, The design of experiments, Cambridge University Press, Cambridge (1988).
[29] J.A. John and M.H. Quenouille, Experiments: Design and Analysis, Griffin, London (1977).
[30] P.W.M. John, Statistical Design and Analysis of Experiments, Macmillan, New York (1971).
[31] K. Shankland, C.J. Gilmore, G. Bricogne, and H. Hashizume, Acta Cryst. A49, 493-501 (1993).
[32] P.E. Nixon and A.C.T. North, Acta Cryst. A32, 325-333 (1976).
[33] J.H. Konnert and W.A. Hendrickson, Acta Cryst. A36, 344-349 (1980).
[34] D.E. Tronrud, L.F. Ten Eyck, and B.W. Matthews, Acta Cryst. A43, 489-501 (1987).
[35] A.T. Brünger, J. Kuriyan, and M. Karplus, Science 235, 458-460 (1987).
[36] G.A. Sim, Acta Cryst. 12, 813-815 (1959).
[37] G. Bricogne, In The Molecular Replacement Method. Proceedings of the CCP4 Study Weekend 31 January – 1st February 1992, edited by W. Wolf, E.J. Dodson and S. Gover, 62-75, Daresbury Laboratory, Warrington (1992).
[38] M. Fujinaga and R.J. Read, J. Appl. Cryst. 20, 517-521 (1987).
[39] E. de La Fortelle and G. Bricogne, Methods in Enzymology, 276, 472-494.
[40] G. Bricogne, In Isomorphous Replacement and Anomalous Scattering, edited by W. Wolf, P.R. Evans and A.G.W. Leslie, 60-68. Daresbury Laboratory, Warrington (1991).
[41] A.R. Kalyanaraman and R. Srinivasan, Z. Kristallogr. 126, 262-??? (1968).
[42] I. Tickle, In Molecular Replacement. Proceedings of the CCP4 Study Weekend 15-16 February 1985, edited by P.A. Machin, 22-26. Daresbury Laboratory, Warrington (1985).
[43] M.F.C. Ladd and R.A. Palmer, Theory and Practice of Direct Methods in Crystallography, Plenum Press, New York (1980).
[44] G. Ardito, G. Cascarano, C. Giacovazzo, and M. Luic, Z. Kristallogr. 172, 25-34 (1985).
[45] G. Cascarano, C. Giacovazzo, M. Luic, A. Pifferi, and R. Spagna, Z. Kristallogr. 179, 113-125 (1987).
[46] F. Pavel ík, J. Appl. Cryst. 22, 181-182 (1989).
[47] F. Pavel ík, Acta Cryst. A46, 869-870 (1990).
[48] B.C. Wang, In Methods in Enzymology, Vol. 115, Diffraction Methods for Biological Macromolecules, edited by H. Wyckoff, C.W. Hirs and S.N. Timasheff, 90-112. Academic Press, New York (1985).
[49] R.E. Dickerson, J.C. Kendrew, and B.E. Strandberg, In Computing Methods and the Phase Problem in X-ray Crystal Analysis, edited by R. Pepinsky, J.M. Robertson and J.C. Speakman, 236-251, Pergamon Press, Oxford (1961).
[50] R.E. Dickerson, J.C. Kendrew, and B.E. Strandberg, Acta Cryst. 14, 1188-1195 (1961).
[51] K.C. Holmes, In Computing Methods in Crystallography, edited by J.S. Rollett, 183-203. Pergamon Press, Oxford (1965).
[52] T.A. Jones and S. Thirup, EMBO J. 5, 819-822 (1986).
[53] D.M. Blow and B.W. Matthews, Acta Cryst. A29, 56-62 (1973).
[54] W.A. Hendrickson and M.M. Teeter, Nature 290, 107-109.
[55] G. Bricogne and J.J. Irwin, in Macromolecular Refinement, edited by M. Moore and E.J. Dodson, 85-92. Daresbury Laboratory, Warrington (1996).
[56] N.S. Pannu and R.J. Read, in Macromolecular Refinement, edited by M. Moore and E.J. Dodson, 75-84. Daresbury Laboratory, Warrington (1996).
[57] G.N. Morshudov, E.J. Dodson, and A.A. Vagin, in Macromolecular Refinement, edited by M. Moore and E.J. Dodson, 93-104. Daresbury Laboratory, Warrington (1996).