The world according to wARP:
improvement and extension of crystallographic phases
Anastassis Perrakis1, Titia K. Sixma1, Keith S. Wilson 2 and Victor S.
Lamzin3
1. Netherlands Cancer Institute (NKI), Department of Molecular Carcinogenesis,
Plesmanlaan 121,
1066 CX Amsterdam, The Netherlands
2. Protein Structure Group, Dept. of Chemistry, University of York,
Heslington, York YO1 5DD, UK
3. European Molecular Biology Laboratory (EMBL) Hamburg, c/o DESY, Notkestrasse
85,
22603 Hamburg, Germany
We have developed procedures for the improvement of crystallographic phases
resulting either from the position of a heavy atom within the native molecule,
or from a multiple isomorphous replacement experiment.
In the first case the position of a heavy atom as located from native Patterson
maps is used as a starting model for least squares or maximum likelihood
refinement and iterative model updating in an ARP procedure. Automatic update
and completion of the model by ARP, results to maps of excellent quality.
Furthermore, the atomic positions of the final ARP model are very accurate and
can be used to initiate automatic model building techniques, currently under
development.
For the second case, the best initial map is used to construct a number of
dummy free atom models which are subjected to ARP refinement. Averaging of the
phase sets calculated from the refined models and weighting of structure
factors by their similarity to an average vector, results in a phase set that
improves and extends the initial phases if the native data set has sufficiently
high resolution (beyond ~ 2.4 Å). This procedure allows shortening of the
time-consuming step of model building in a lot of crystallographic structure
solutions.
NOTE: The ARP program is freely available as part of the CCP4
package. C-shell scripts and the actual averaging program, are available to run
wARP. They perform the dummy model building, ARP refinements and final
averaging in an automated manner. They are also capable to split jobs in a
`parallel' manner to different processors which can be located in different
computers over a network, thus minimizing the actual required run time to the
one needed for a single ARP job - provided that enough processors are
available. The scripts are tested on several Irix 5.3 based clusters, but
should be straight-forward to adapt for usage with any Unix based system. A WWW
ARP/wARP home page is now available, at http://den.nki.nl/~perrakis/arp.html
from where the complete ARP/wARP package can be obtained. A mailing list is
also open for questions and discussion for ARP/wARP usage. To subscribe, simply
do it through the WWW page or send a mail with one line `subscribe arp-users'
to majordomo@linde.nki.nl.
One single or a few heavy atoms located from the native Patterson synthesis are
used as an initial model. Starting from these atoms, a model consisting of
only oxygen atoms is slowly created by ARP. This model consists of free atoms
that are not subjected to any kind of restraint. One ARP refinement cycle has
two parts: (1) unrestrained least-squares minimization or maximum likelihood
refinement in reciprocal space, to properly match calculated to observed
structure factor amplitudes and (2) substantial modification of the current
atomic dummy model in real space, using ARP [1,2]. For the unrestrained
refinement step, C-shell scripts have been constructed to employ most currently
available programs in the procedure. Standard protocols include PROLSQ [3] and
REFMAC [4] from the CCP4 [5] suite. ARP, after each reciprocal space refinement
cycle, updates the model mimicking human intervention between refinement
cycles. It removes atoms based on the density in the 3Fo-2Fc Fourier synthesis
and adds atoms in significant density in the Fo-Fc Fourier synthesis, provided
that they are bonded to existing atoms. After several such cycles of ARP, the
atoms that are added gradually constitute a model that resembles the protein to
a great extend.
The procedure described above requires data to very high resolution to be
available and a heavy atom present in the native protein. In most
crystallographic projects, however, this is not the case. Since it is very
hard if at all possible to provide an ab initio solution to the phase
problem in such cases, our effort has been concentrated on improving phases
that are available by experimental techniques. Such phase information can be
very inaccurate and means of improvement will speed up the efficiency and the
quality of model building. ARP needs high resolution data to converge to
global minimum during refinement. If such data are not available, the
refinement will most likely not converge and inaccuracies are introduced to the
`final' model. With wARP we try to overcome this problem by the weighted
averaging of structure factors from individual models.
The first step in the wARP procedure is the creation of moderately different
free atom models in the best available map. The procedure for building a `dummy
model' is then invoked as described in the ARP manual. Briefly, starting from a
small set of dummy atoms placed anywhere in the protein region, a model is
slowly expanded by the stepwise addition of atoms that are in bonding distances
with existing atoms and in significant density in the electron density map
exists for their placement. Six such models are created, using slightly
different ARP building protocols, which are used for all subsequent steps.
Next, these models are subjected to ARP refinement. Due to the limited amount
of diffraction data, they will presumably at the end contain different errors,
which by the averaging procedure will be canceled out.
Structure factors are calculated for all models after refinement and scaled to
observed amplitudes. A vector average of the calculated structure factors from
the different refined models is then calculated. The phase of the vector
average is remarkably better than those calculated from any of the individual
runs. Subsequently, a weighting scheme is applied to enhance the overall
quality of phases, depending on the variance of the individual structure
factors around the average.
Rubredoxin is a small protein of 51 residues, the first protein to be refined
to atomic resolution [6]. It contains a Fe atom which is coordinated by 4 Cys
residues. The position of the Fe atom and the 4 sulfurs of the cysteines side
chains can be located from the native Patterson map, if data better than 1.5
Å resolution are available. A high resolution data set of rubredoxin
(0.92 Å) was used. In all cases that lower resolution is quoted, that
means that the data were simply truncated at that resolution limit.
The starting model for the ARP refinement procedure was initially the Fe atom
with the coordinates calculated from the native Patterson map. After 80 cycles
of least squares refinement, or 30 cycles of maximum likelihood refinement a
complete model was available. The map correlation coefficient [7] improved
from only 26 % to more than 90 % in both cases. The lowest resolution at which
the method works, starting from the Fe atom alone, is 1.1 Å. However, if
the positions for the four sulfur atoms are included, the method can work with
1.4 Å data, in other words with less than one third of observed
reflections at 0.92 Å. In that case, the correlation coefficient with
the final map is 96%, because the correct atomic types are used for the four
sulfur atoms. If we try to use data to 1.5 Å resolution, there is a
small increase in correlation coefficient to 45%, but after that no improvement
could be achieved. Protocols involving the use of E-maps and the wARP
averaging described below are being tested to extend the use of method to much
lower maximal resolution. It is of interest to note, that the atomic positions
of atoms placed by ARP are these of atoms in the final model, with slight
variation, Figure 1. It would be thus feasible to use them to initiate
automatic model building techniques to minimize the amount of time spent in
traditional model building and the errors introduced by this procedure.
Characteristic parts of the maps before and after the ARP procedure are shown
in Figures 2 and 3.
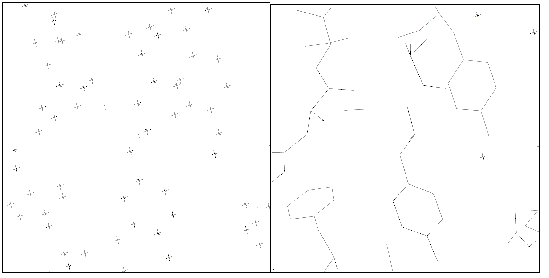
Figure 1
Positions of the ARP atoms (left) and of the atoms in the final model (right),
for a representative region of the protein. You are welcome to try the `join
the correct dots' game in the left panel of the figure ...
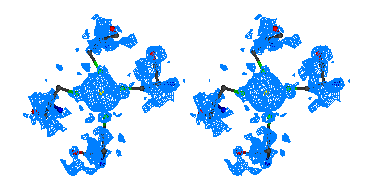
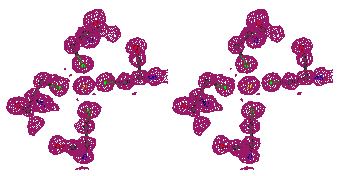
Figure 2
Stereo figures of the area of the map around the starting Fe atom. In the
initial map (top) resulting from the phases calculating from Fe atom position
alone, a big bolb of density is representing the ion. Although there is
density for the four sulfurs of the cysteines coordinating the Fe ion, it is
hardly interpretable. After ARP the atomic positions are clearly visible and
the map is of excellent quality.
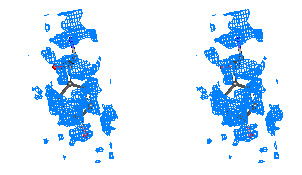
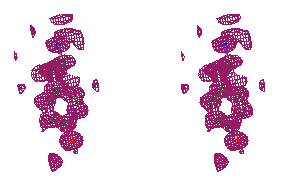
Figure 3
Stereo figures of one area of the map far from the starting Fe atom. In the
initial map (top), although density for some of the Tyr atoms is present, the
Tyr residue is in practice not recognizable. After ARP refinement the Tyr main
and side chains are clearly recognizable.
The structure of the Leishmania virus coat protein (Leishmanolysin, PSP)
was solved with a complicated protocol involving the use of SIRAS phases for
two different crystal forms, averaging between those, solvent flattening and
density skeletonization (unpublished data were kindly provided by Dr. Peter
Metcalf). For the wARP test one set of SIRAS phases was used, which extends to
a resolution of 3.0 Å. These phases were determined for the first crystal
form for which native data extending to 2.5 Å were used for solvent
flattening and phase extension with the DM program [16], CCP4. This density
modified map was used to build the initial models with ARP. The ARP
unrestrained refinement was performed against a higher resolution native data
set from a frozen crystal (2.0 Å). REFMAC maximum likelihood minimization
was used with ARP. All of these models gave maps of dramatically better quality
than the solvent flattened map. Here the power of ARP procedure itself is large
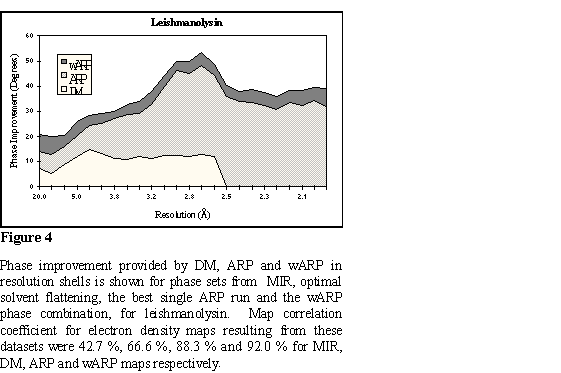
because the resolution of the native data is good. The wARP procedure resulted
in a small but significant additional improvement. Statistics on phase
improvement are in Figure 4 and a representative part of the map at Figure 5.
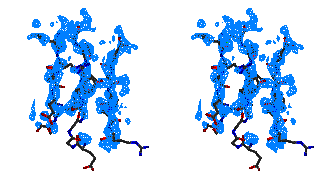
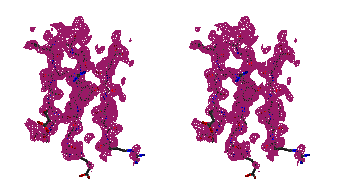 Figure 5
Figure 5
Representative
regions of the solvent flattened (a,c) and equivalent wARP averaged maps (b,d)
for Leishmanolysin, shown in stereo.
The chitinase A structure from Serratia marsescens (ChiA) was initially
solved by MIRAS [8]; with one only derivative contributing to better resolution
than 5.0 Å [ref]. The MIRAS map (2.5 Å) was solvent flattened with
the procedures implemented in the PHASES package [15]. Model building was not
straightforward and much time was spent in tracing the protein chain. In the
wARP procedure the solvent flattened map was used to initiate building of dummy
models. PROLSQ least squares minimization against the native 2.3 Å data
was used with ARP. Refinement of the models resulted in crystallographic R
factors ranging between 20.1 % and 22.4 %. All of the ARP refined models gave
phases same or worse than the phases already available by solvent flattening,
due to the limited resolution of the native data, Figure 1a. At that case,
where limited resolution if the data prevent convergence of the refinement, the
wARP averaging procedure results in a much further improved map, comparable to
the improvement achievable with higher resolution data. The phase improvement
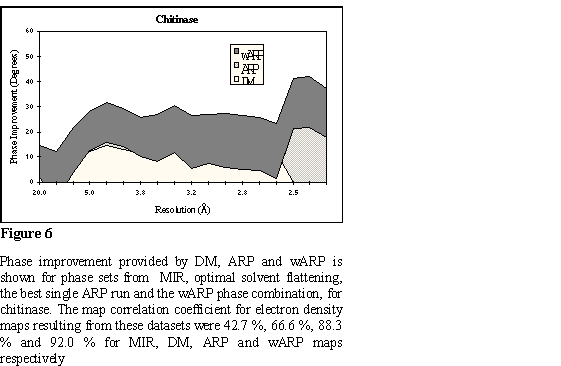
in resolution shells, for all phase sets, is analytically shown in Figure 6. A
characteristic region of the map is shown in Figure 7.
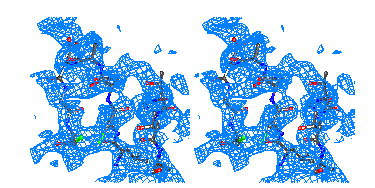
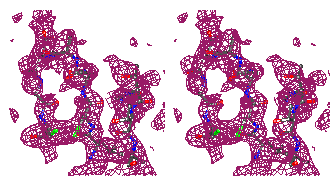 Figure 7
Figure 7
Representative
regions of the solvent flattened (a,c) and equivalent wARP averaged maps (b,d)
for Chitinase A, shown in stereo.
Ab initio methods in protein crystallography have only recently been
successfully applied, the most characteristic examples being the structure
solution of crambin [9] by direct methods and cytochrome c6 [10] by Patterson
expansion methods. The limitation for succesful application of ab initio
methods is the resolution of the diffraction data. Allthouh our current
example, rubredoxin, can be solved easily by any relevant procedure if atomic
resolution data are available, these methods fail if data worse than ~ 1.2
Å are available. With ARP we managed to produce an excellent map and an
atomic model, with only 1.4 Å data, ie with essentially ~ 60 % of the
reflections. Many more proteins diffract to resolution around 1.5 Å than
1.2 Å, according to the data on projects recently collected at EMBL
Hamburg synchrotron X-rays facilities. Furthermore, we believe that we will be
able to extend that limit in the near future, possibly with the application of
wARP averaging.
In contrast to most density modification methods the wARP procedure is
extremely sensitive to the resolution of observed data in the native dataset.
This is due to the limitations of the unrestrained refinement step, which
requires that the observations/parameters ratio is more than 1.5 for
convergence to a minimum. It is crucial to realise, that the real limitation
can not be expressed solely in resolution terms, but better as
observations/parameters ratio, which is largely dependent on solvent content.
Thus, for a crystal with high solvent content 2.5 Å data will be
sufficient while for a crystal with low solvent content data to 2.0 Å
resolution must be available. Obviously the collected data must be of good
quality, as can be judged by Rmerge, I/ (I), and completeness. The
success of refinement can be easily assessed by the crystallographic R
factor.
(I), and completeness. The
success of refinement can be easily assessed by the crystallographic R
factor.
Our experience shows that if the ratio of the number of reflections in the
dataset to refined atomic parameters (four parameters per atom, x,y,z,B)
is more than 2.0 (resolution ~ 2.0 Å) then use of maximum likelihood
refinement as implemented in REFMAC can be used very effectively, as shown in
Leishmanolysin. If the observations to parameters ratio drops below 2.0
traditional least squares refinement as implemented in PROLSQ produce better
results, as shown for ChiA. When the observations to parameters ratio drops
below 1.5 the method does not work.
The averaging method we describe has also been succesfully used in our
laboratory to combine maps obtained by different phasing techniques. We have
used MIR phase sets determined for `cold' and `warm' native datasets, different
solvent flattening protocols and partial model phase sets, to combine them with
the wARP procedure. The resulting map appears to be of substantially better
quality. Unfortunately, this project is still under refinement and we can not
quote the exact phase improvement figures. Furtermore, it is not a usual case
to obtain many phase sets, with different sources of errors. Also, other more
standard and theoretically sound procedures are developed for standard phase
combination. Thus, we will not treat it as a test case, allthough potential
users that think this procedure might be applicable in their particular cases
are encouraged to inquire after this possibility with us.
1. Lamzin, V.S. & Wilson, K.S. (1993) Automated refinement of protein
models. Acta Crystallogr. D49, 129-147.
2. Lamzin, V.S. & Wilson, K.S. (1996) Automated refinement for protein
crystallography. In Methods Enzymol.: Macromolecular Crystallography.
(Carter, C.M. & Sweet, R.M. Eds.) in the press
3. Konnert, J.H. & Hendrickson, W.A. (1980) A restrained-parameter
thermal-factor refinement procedure. Acta Crystallogr. A36,
344-350.
4. Murshudov, G., Vagin, A. and Dodson, E (1996) Application of maximum
likelihood refinement. In The refinement of protein structures
Proceedings of Daresbury study weekend
5. CCP4 (1994) Collaborative Computational Project Number 4. The CCP4 suite:
programs for protein crystallography. Acta Crystallogr. D50,
760-763.
6. Dauter, Z., Sieker, L.C. & Wilson, K.S. (1992) Refinement of rubredoxin
from Desulfovibrio vulgaris at 1.0 Å with and without restraints. Acta
Crystallogr. B48, 42-59.
7. Watenpaugh, K.D., Sieker, L.C. & Jensen, L.H. (1980) Crystallographic
refinement of rubredoxin at 1.2 Å resolution. J. Mol. Biol. 138,
615-633.
8. Lunin, V.Y. & Woolfson, M.M. (1993) Mean phase error and the map
correlation coefficient. Acta Crystallogr. D49, 530-533,
9. Cowtan, K. (1994), Joint CCP4 and ESF-EACBM Newsletter on Protein
Crystallography, 31, 34-38
10. Perrakis, A.,et al and Vorgias, C.E. (1994) Crystal structure of a
bacterial chitinase at 2.3 Å resolution. Structure 2,
1169-1180.
11. Furey, W & Swaminathan, S.(1990). PHASES - a program package for the
processing and analysis of diffraction data from macromolecules. American
Crystallographic Association Meeting Abstracts, 18, 73
12. Weeks, C.M., Hauptman, H.A., Smith, G.D., Blessing, R.H., Teeter, M.M.
& Miller, R. (1995) Crambin: a direct solution for 400-atom structure. Acta
Crystallogr. D51, 33-38.
13. Frazao, C., Soares, C.M., Carrondo, M.A., Pohl, E., Dauter, Z., Wilson,
K.S., Hervas, M., Navarro, J.A., De la Rose, M.A. & Sheldrick, G.M. (1995)
Ab initio determination of the crystal structure of cytochrome c6
and comparison with plastocyanin. Structure 3,