Contents
Load homologous proteinsSuperpose by secondary structure matching
Superposing binding sites
Superposing the ligand
Multiple homologous chains
See general Superpose documentation
 |
CCP4 Molecular Graphics Tutorials | |
| Superpose Proteins |
| Documentation Contents | On-line Documentation | Tutorials | CCP4mg Home |
Select two homologous proteins such as 1df7 and 4dfr (hint to remove existing models: from the Edit menu select For all data and Close).
To set a suitable colour and display style for all models open the Picture wizard window from the Applications menu. In the picture wizard window select colour molecule from the bonds folder in the list at the top of the window. Click the Create picture button to redraw the scene. (Note that the Picture Wizard changes are applied to all visible models and by default any existing display objects are deleted.)
From the Applications pull-down menu select Superpose.
In the new window check that the Superpose mode (at the top of the
window) is Secondary structure matching(SSM) and hit the Superpose button. The models will
be superposed. To show the equivalent residues and distances between CA atoms check the box labelled Show matchs with...
In the Models to superpose frame of the interface is a column labelled Show match. After an SSM superposition the column will contain comboboxes for each moving models. The 4dfr structure contains two equivalent chains and the SSM algorithm finds alternative possible matches for both chains which you can review by stepping though the options in the combo box in the Show match column.
The SSM method gives a good global superposition but sometimes localised regions can be better superposed by matching only a selection of residues in the region. A better local superposition can particularly help in comparing binding sites. Selecting the appropriate residues to superpose may require some time and thought but the Match close residues method is a simple automatic method that is worth a try. The method requires that the models are already roughly superposed and that a limited set of residues are selected in the FIXED model. The algorithm matches residues in the moving model that are close to the selected residues in fixed model and superposes them.
The 1df7 and 4dfr structures both contain the ligand MTZ. To
optimise superposition of the MTZ binding site select the Match close
residues method and then for the fixed model select the MTZ
binding site by clicking the model selection widget lassoo ![]() and pick
Neighbourhood.. from the menu to open a new window.
and pick
Neighbourhood.. from the menu to open a new window.
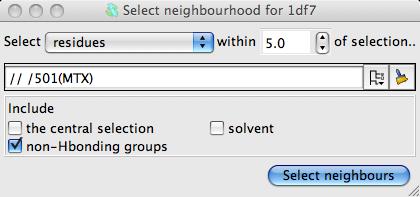
In the new window select the MTZ ligand as the centre of the neighbourhood and click the Select neighbours botton. This window closes and the seelction command appears in the main Superpose window. You can see which residues have been selected by checking the box labelled Highlight selected atoms. Now click the Superpose button to superpose the close residues. There will be only a slight movement to optimise the superposition of the binding site.
This part of the tutorial introduces the tools for manually defined matches and uses these tools to superpose equivalent ligands.
Click on the User defined matches method. The window will change significantly to show a User defined matches frame.
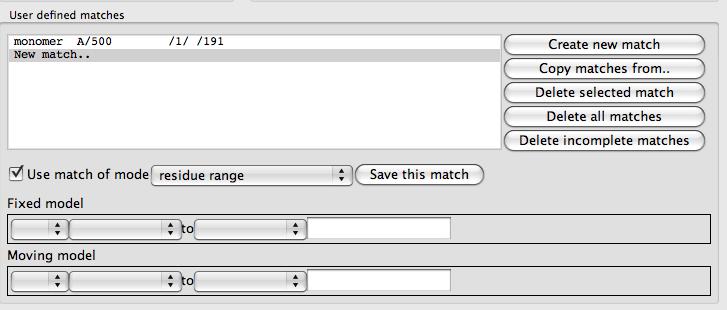
At the top of this frame is a list box of the defined matches that is initially empty except for a New match.. option. To the right of the list is a set of buttons with tools to edit the match list. Beneath the list are tools for specifying one match. There are various modes for a match: atom, residue, residue range, monomer etc. and the selection widget will be changed appropriately for each match mode. From the combo box labelled match mode select monomer. Use the selection widgets to select the MTX ligand for 1df7 (// /501[MTX] - beware the chain does not have a name) and 4dfr (A/160[MTX]). You must click the Save this match button to get this match saved to the list. The matches between the two MTX molecules should be shown on the display. You can now click Superpose to superpose the two ligands.
The matching algorithm for monomers is capable of finding the equivalent atoms in non-identical molecules. It uses a graph matching approach which looks at the atom element types and the bonds between the atoms. It takes no account of the atom names.
Further matches can be added using the interface or they can be selected by clicking on atoms in the model with right mouse button and using the Superpose options which appear at the bottom of the popup menu. Try using the menu item Superpose - select this -> Residue to select an active site residue then click on the equivalent residue in the other model and the menu should now include Superpose - match this to with a list of unmatched items in the other model.
Matches selected by clicking on the model are shown in the match list and can be edited. A possible useful feature is the ability to switch off a match: click on the match in the list to make it the edited match then uncheck the box labelled Use match and remember to click the Save match button. The inactive matches are shown greyed-out in the match list.